Resumo
Apesar do crescente interesse por produtos farmacêuticos derivados da Cannabis sativa L., a maioria das alegações terapêuticas relacionadas à espécie carece de evidências científicas robustas, o que prejudica o seu uso racional. Outros fatores, como a inequidade de acesso aos produtos disponíveis, a assimetria de informação e o estigma social em torno da espécie, impõem desafios adicionais à regulação desses produtos. O modelo regulatório brasileiro tem-se desenvolvido ao longo da última década, destacando-se a publicação da RDC 327/2019, que possibilitou a Autorização Sanitária de produtos de Cannabis para fins medicinais, ampliando as alternativas disponíveis aos pacientes não responsivos a outros tratamentos. Existem, porém, desafios a serem superados e oportunidades de aprimoramentos, conforme apontado pelo estudo de Análise de Impacto Regulatório (AIR) conduzido pela Agência Nacional de Vigilância Sanitária (Anvisa), com relatório recentemente publicado. Espera-se que o avanço de discussões sobre o tema, não apenas no âmbito da Anvisa como em outras esferas, contribua para a evolução do modelo atual, com base no conhecimento científico existente, buscando atender às necessidades da população.
Palavras-chave Regulamentação governamental; Política de saúde; Lacunas de evidências; Saúde pública
Abstract
Despite the growing interest in pharmaceutical products derived from Cannabis sativa L., most of the therapeutic claims related to the species lack robust scientific evidence, which hinders rational use. Other factors, such as the unequal access to available products, the asymmetry of information, and the social stigma surrounding the species, impose additional challenges for regulators. The Brazilian regulatory model has evolved over the last decade, most notably with the publication of RDC 327/2019, which enabled the Sanitary Authorization of cannabis products for medicinal purposes, expanding the alternatives available to patients unresponsive to other treatments. However, there are still challenges to overcome, as well as improvement opportunities, as pointed out by the Regulatory Impact Analysis (RIA) study conducted by the National Health Surveillance Agency (ANVISA), whose report was recently published. It is expected that the progress of the discussions on the subject, not only by Anvisa but also in other spheres, will contribute to the progress of the current model, based on available scientific knowledge, aiming to meet the population's needs.
Keywords Medical Cannabis; Government Regulation; Health Policy; Evidence Gaps; Public Health
Contextualização
O uso de produtos obtidos a partir de Cannabis sativa L. com finalidades medicinais não é um fenômeno propriamente novo, tendo sido relatado em diferentes civilizações desde a antiguidade[1,2]. No entanto, foi apenas a partir das últimas décadas do século XX que a evolução do conhecimento acerca dos constituintes da espécie e de suas características farmacológicas abriu caminho para o desenvolvimento de medicamentos com base científica[1,2]. Contudo, prevalecem algumas limitações de acesso à espécie para fins de pesquisa, o que dificulta a superação de lacunas de conhecimento existentes[1,3].
O aumento do interesse pela exploração terapêutica de C. sativa tem se refletido em uma demanda crescente de acesso a seus produtos, embora nem sempre alinhada ao estado da arte do conhecimento científico[1,3]. A maioria das alegações terapêuticas atribuídas à espécie ou seus constituintes carece de evidências de eficácia robustas[4,5] e o conjunto de produtos contendo esse tipo de insumo que passaram por desenvolvimento clínico adequado para demonstração de seu benefício terapêutico em condições específicas é limitado, o que se reflete em número reduzido de medicamentos registrados [1,3].
Nesse contexto, a regulação desses produtos tem sido objeto de debate em diferentes países, resultando no desenvolvimento de modelos diversos, refletindo os contextos locais[1,2]. Um desafio desse processo é desenvolver estratégias que não apenas busquem lidar com as demandas de parte da população por acesso aos produtos, mas também envolvam mecanismos para controle adequado de riscos e promoção do uso racional e baseado em evidências[2,3]. Somam-se, ainda, aspectos que perpassam as questões regulatórias, como a inequidade de acesso, a assimetria de informação e os impactos do estigma social em torno da espécie sobre a percepção pública e as escolhas de pacientes e prescritores[1-3,6]. Trata-se, portanto, de um problema complexo que demanda abordagens de enfrentamento multidisciplinares e intersetoriais.
Este artigo busca apresentar uma visão geral do marco regulatório brasileiro, discutindo os principais desafios no cenário atual e seus impactos no processo de tomada de decisão regulatória, bem como possíveis estratégias e perspectivas para aprimoramentos futuros.
Evolução do marco regulatório brasileiro nas últimas décadas
O Estado brasileiro também se insere, com seu papel regulador, no contexto global de crescente oferta e demanda por produtos farmacêuticos obtidos de C. sativa. Desde 2014, a Agência Nacional de Vigilância Sanitária (Anvisa) tem desenvolvido estratégias para regulação desses produtos[3].
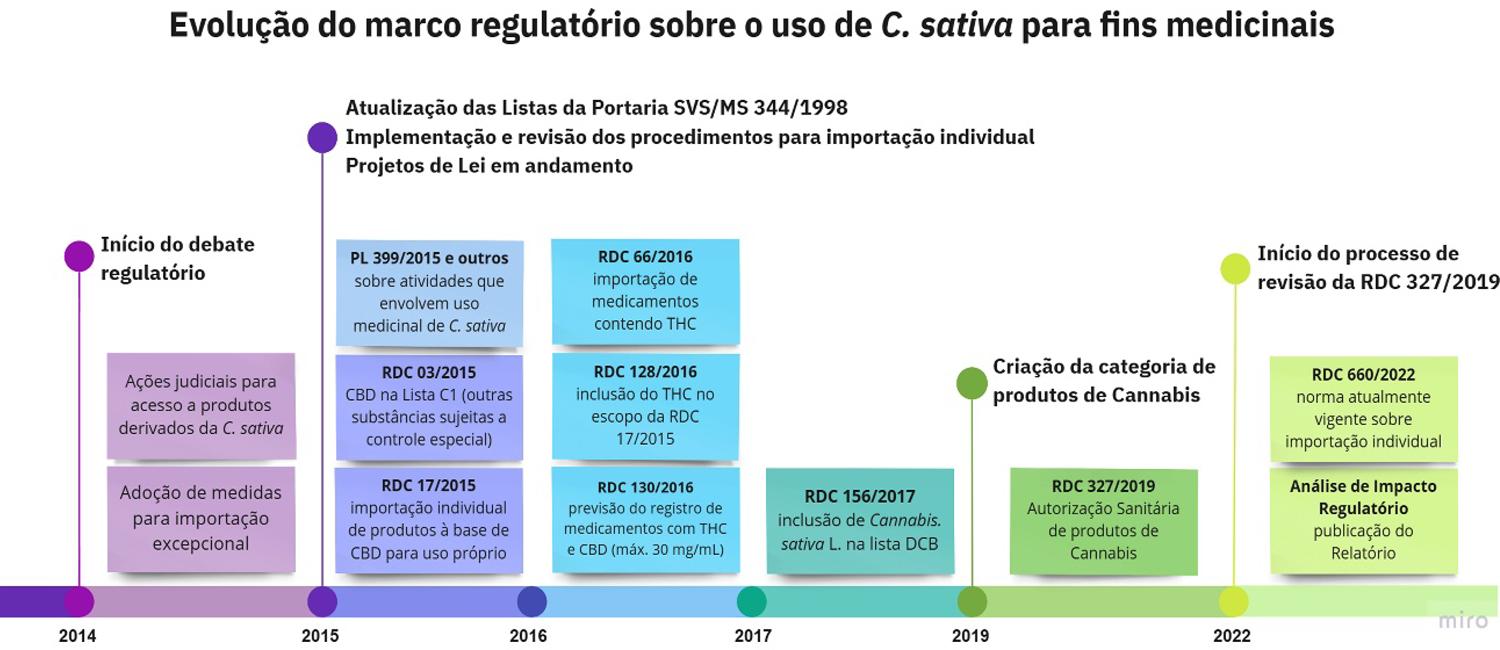
A Portaria MS/SVS 344/1998 lista as substâncias sujeitas ao controle especial no Brasil. A Cannabis sativa L. consta atualmente na Lista E (plantas proscritas e seus derivados); enquanto o tetraidrocanabinol (THC) está na Lista F2 (substâncias psicotrópicas de uso proscrito). Em 2015, o canabidiol (CBD) foi incluído na Lista C1 (outras substâncias sujeitas a controle especial), sendo excetuado dos controles previstos para substâncias obtidas a partir das espécies da Lista E. No mesmo ano, foi publicada a RDC 17/2015, que permitiu a importação por pessoa física, para uso terapêutico próprio, de produtos à base de CBD, mediante prescrição. Em 2016, o THC foi incluído no Anexo I dessa norma e uma atualização da Portaria 344/1998 permitiu o controle de medicamentos contendo THC e CBD em concentrações máximas de 30 mg/mL (Lista A). Em 2017, foi registrado o medicamento Mevatyl®, contendo THC (27 mg/mL) e CBD (25 mg/mL), indicado para o tratamento da espasticidade relacionada à esclerose múltipla.
A RDC 17/2015 foi revisada em 2019, 2020 e 2021 e, atualmente, os procedimentos para importação individual de produtos derivados da Cannabis estão estabelecidos pela RDC 660/2022.
Após discussões técnicas, a Anvisa publicou a RDC 327/2019, que criou a categoria regulatória de produtos de Cannabis e estabeleceu requisitos para sua Autorização Sanitária (AS), em caráter transitório, possibilitando que sejam acessíveis mediante prescrição. A evolução do cenário regulatório está ilustrada na FIGURA 1.
: Linha do tempo com a evolução do debate regulatório referente ao uso de produtos derivados da C. sativa para fins medicinais. Elaborada pelas autoras utilizando a plataforma Miro®.
No âmbito do Congresso Nacional, estão em tramitação Projetos de Lei (PL) relacionados à regulação de atividades envolvendo aplicações industriais e medicinais de C. sativa. Entre estes, cita-se o PL 399/2015, que propõe regulamento do plantio da espécie, bem como da fabricação e comercialização de medicamentos que contenham insumos ativos dela obtidos. Além disso, recentemente, o Superior Tribunal de Justiça (STJ) validou a possibilidade de autorização de cultivo de variedade de C. sativa com baixo teor de THC por pessoas jurídicas, para fins exclusivamente medicinais e farmacêuticos.
Principais características do modelo regulatório atual
Há diferentes vias legais de acesso no Brasil aos produtos derivados da C. sativa com finalidade medicinal: importação por pessoa física, aquisição de medicamento regularmente registrado e de produtos de Cannabis autorizados conforme RDC 327/2019. Existem, ainda, as permissões obtidas em caráter excepcional mediante ações judiciais[3].
A categoria regulamentada pela RDC 327/2019 contempla os produtos farmacêuticos industrializados contendo CBD ou extratos da C. sativa como princípios ativos destinados a pacientes refratários a outras terapias disponíveis no país. Os produtos contendo THC em concentrações superiores a 0,2% são sujeitos a restrições diferenciadas, destinados a cuidados paliativos de pacientes em situações clínicas irreversíveis ou terminais, um racional análogo ao do uso compassivo de medicamentos. A indicação e o esquema posológico são de responsabilidade do prescritor.
A autorização desses produtos não é condicionada à comprovação de eficácia, entretanto, são avaliados aspectos de qualidade e sua obtenção deve cumprir as Boas Práticas de Fabricação de medicamentos.
A transitoriedade dessa categoria se traduz na validade limitada da AS, de 5 anos. Após esse período, o produto deve obter registro definitivo como medicamento para continuar a ser comercializado no país. Para tanto, devem ser apresentadas evidências de eficácia adequadas.
Desde 2020, foram autorizados cerca de 40 produtos de Cannabis (consulta disponível em: https://consultas.anvisa.gov.br/#/cannabis/), possibilitando, assim, a ampliação das alternativas terapêuticas disponibilizadas nacionalmente.
Caracterização do problema e proposição de estratégias regulatórias
As decisões relacionadas à regulação de produtos farmacêuticos, inclusive aqueles obtidos a partir de C. sativa com finalidade terapêutica, devem ser equilibradas de forma a garantir prioritariamente a segurança dos pacientes, sem criar barreiras de acesso desproporcionais, e buscando, ainda, construir um cenário favorável ao desenvolvimento de atividades de pesquisa e inovação[1-3].
Nesse contexto, as Análises de Impacto Regulatório (AIR) são ferramentas úteis para a elaboração dessas estratégias considerando seus efeitos sobre a saúde pública[3]. A AIR segue abordagens sistemáticas, embasadas em ciência, para fundamentar decisões regulatórias, para que sejam adotadas medidas eficazes, eficientes e proporcionais aos riscos que pretendem mitigar.
Considerando o caráter temporário da RDC 327/2019, conforme seu art. 77, a Anvisa iniciou a sua revisão em 2022. Durante o processo, foi conduzido um estudo de AIR, com participação de representantes de diversas áreas da Agência, incluindo as autoras deste artigo, sendo o Relatório publicado em 05/2024[3]. A metodologia utilizada envolveu a definição do problema objeto da análise, suas causas e consequências; o delineamento dos objetivos; o levantamento de possíveis alternativas de atuação regulatória e de seus possíveis impactos[3].
O problema regulatório foi definido à época como “Dificuldades em atender às demandas de parte da população por produtos medicinais à base de Cannabis que supram as suas necessidades terapêuticas de forma racional”[3]. Os principais fatores apontados como causas ou consequências do problema e suas inter-relações são sintetizados a seguir.
Estigma: o estigma construído em torno da C. sativa, reforçado pela criminalização do seu uso, influencia a opinião pública e as decisões regulatórias e políticas, dificultando a revisão de medidas de controle. Como consequência, observou-se historicamente um cenário pouco favorável às iniciativas de pesquisas científicas, tanto pela tendência de se evitar a condução de estudos com uma espécie considerada proscrita quanto pelas dificuldades de acesso decorrentes dos controles aplicados[3].
Insuficiência de evidências científicas e seus efeitos sobre a racionalidade e uso dos produtos: a diversidade de quimiotipos de C sativa, com variadas proporções entre canabinoides com perfis farmacológicos complexos e distintos entre si, prejudica a consolidação de resultados de diferentes estudos, dadas as diferenças entre os materiais avaliados em cada caso, que inviabilizam extrapolações e comparações diretas. Além disso, não há caracterização adequada de muitos dos produtos estudados, o que dificulta ainda mais o processo de síntese e delineamento de conclusões a partir do conjunto de evidências. Esses fatores somam-se às falhas metodológicas e vieses de muitos dos estudos disponíveis como dificultadores da construção de evidências de segurança e eficácia sólidas para a maioria das alegações terapêuticas atribuídas à espécie[1,3,4]. Cabe mencionar, ainda, que os produtos de Cannabis autorizados nos termos da RDC 327/2019 não têm indicações definidas e não há uma lista de condições diagnósticas elegíveis previamente estabelecidas, o que coloca a responsabilidade dessas definições sobre os prescritores[3]. Situação análoga ocorre para outros produtos não regularizados como medicamentos, a exemplo daqueles importados por meio de autorizações individuais e dos obtidos de associações ou autocultivo. Diante da ausência de diretrizes terapêuticas claras, o uso racional desses produtos fica comprometido[3]. Adicionalmente, a ausência de exigências regulatórias de evidências de segurança e eficácia para os produtos de Cannabis pode levar percepções enviesadas quanto à necessidade de condução de estudos clínicos envolvendo tais produtos, o que contribui para a perpetuação de lacunas de conhecimento e compromete a construção de evidências científicas robustas para embasamento do registro de novos medicamentos[1-3].
Falta de equidade no acesso aos produtos: de forma geral, os preços dos produtos farmacêuticos obtidos da espécie, incluindo os regularizados como produtos de Cannabis e aqueles importados mediante autorizações individuais, são elevados, impactando negativamente o acesso dos pacientes a essas alternativas[1,3]. Esses produtos não são passíveis de controle de preço por não serem enquadrados como medicamentos, o que contribui para esse cenário. Além disso, fatores que impactam nos custos de produção, como a falta de regulamentação do cultivo da espécie no país e, consequentemente, de autonomia para obtenção de insumos ativos, também contribuem para o aumento do custo final dos produtos[1,3]. Os impactos em termos de saúde pública são ainda mais graves se consideradas as demandas judiciais para custeio desses tratamentos pelo Sistema Único de Saúde (SUS), inclusive de produtos importados que não passaram por regularização no Brasil e comumente não são registrados como medicamentos nos países de origem[1,3,6]. Em um cenário em que os recursos públicos são conhecidamente limitados, a destinação de recursos para custeios de tratamentos sem a demonstração de uma relação risco-benefício favorável pode ser problemática[1,3,6]. Outra consequência da dificuldade de acesso a produtos industrializados é a busca de estratégias alternativas como o autocultivo. Cabe observar que os produtos obtidos em escala artesanal não passam por controles que garantam a padronização e reprodutibilidade de suas características, resultando em riscos de efeitos indesejados devido à presença de determinados canabinoides em níveis inadequados e variabilidade dos efeitos ao longo do tempo[1,3].
Divulgação de informações falsas e distorções de percepções na formação da opinião pública: a divulgação de informações que subestimam os riscos e supervalorizam supostos benefícios de produtos obtidos de C. sativa, sem embasamento em evidências, contribui para a construção de demandas irrealistas e prejudicam o uso racional[1,3]. Muitos dos responsáveis pela divulgação dessas informações são agentes movidos por interesses econômicos, nem sempre compatíveis com a proteção da saúde[3,6]. Nota-se, ainda, que a implementação, por alguns países, de modelos regulatórios permissivos em relação ao acesso à C. sativa para fins terapêuticos tem motivado pressões sociais pela adoção de estratégias semelhantes no contexto brasileiro[3]. Contribuem para isso as dificuldades de compreensão das limitações dessas estratégias e dos riscos relacionados à adoção de critérios menos rigorosos em termos de qualidade, segurança e eficácia dos produtos de Cannabis, principalmente quando há dificuldades de monitoramento pós-comercialização.
Considerações finais e perspectivas
A regulação de produtos obtidos de C. sativa com finalidade terapêutica relaciona-se a um conjunto complexo de fatores, incluindo aspectos socioeconômico-culturais e de saúde pública. Nos tempos da pós-verdade, é importante considerar, ainda, a disponibilidade e capacidade de interpretação de informações no campo das ciências da saúde[1,3]. Assim, um dos maiores desafios da regulação é alinhar expectativas e demandas de acesso aos produtos às evidências de segurança e eficácia existentes, fornecendo ferramentas para tornar clara a relação benefício-risco envolvida[1,3,6]. Para tanto, é importante que os processos de tomada de decisão sejam conduzidos de forma racional e embasados em evidências[1-3].
A publicação da RDC 327/2019 representou avanços na regulação de produtos farmacêuticos obtidos de C. sativa, que devem atender a critérios de qualidade definidos, possibilitando o seu acesso por pacientes não responsivos a outros tratamentos. O caráter transitório da AS também representa um incentivo à construção de evidências que possibilitem a regularização definitiva dos produtos como medicamentos.
Entretanto, apesar das evoluções regulatórias recentes, há ainda desafios para superação das causas do problema identificado durante o estudo de AIR[3]. Nesse sentido, o relatório indica alternativas regulatórias envolvendo aprimoramentos da RDC 327/2019 e estratégias complementares, que subsidiaram a construção da minuta normativa disponibilizada para contribuições por meio da Consulta Pública 1.316/2025. Entre as alterações propostas destacam-se a inclusão de novas vias de administração permitidas; o detalhamento de procedimentos para importação de insumos para pesquisa, desenvolvimento e fabricação de produtos de Cannabis; a revisão do tipo de receituário especial para produtos com até 0,2% de THC; a perspectiva de manipulação de produtos à base de CBD purificado, apenas; e a possibilidade de renovação da AS, uma única vez, condicionada à demonstração de que o desenvolvimento clínico do produto foi iniciado. Esta condição poderá incentivar a construção de evidências científicas conclusivas para subsidiar o registro definitivo desses produtos como medicamentos. Ressalta-se, porém, que a minuta ainda pode sofrer algumas alterações após a consolidação da CP.
Adicionalmente à atuação da Anvisa, a regulamentação de aspectos importantes para a construção do modelo regulatório nacional depende de outras esferas. Nesse cenário, a discussão dos Projetos de Lei relacionados ao tema no Congresso Nacional pode contribuir para a evolução do modelo atual[1]. Em 13/11/2024, o Superior Tribunal de Justiça (STJ) publicou uma decisão em que considera juridicamente viável a concessão de autorização a pessoas jurídicas para cultivo e comercialização de C. sativa com teor de THC inferior a 0,3% no Brasil, com finalidade farmacêutica. A regulamentação dessa atividade será de responsabilidade da Anvisa e da União, no âmbito de suas competências. Com isso, há a perspectiva de que seja possível obter insumos farmacêuticos a partir da espécie a um custo menor, contribuindo para pesquisa desenvolvimento e fabricação nacional de produtos. Por fim, importa ressaltar a necessidade de aprimoramento contínuo do modelo, considerando a evolução do conhecimento científico, os desafios de saúde pública e as necessidades da população[1].
Referências
- 1 Souza MR, Henriques AT, Limberger RP. Medical cannabis regulation: an overview of models around the world with emphasis on the Brazilian scenario. J Cannabis Res [Internet]. 2022 Jun 16 [cited 2022 Jun 20]; 4(1): 33. Available from: [ https://jcannabisresearch.biomedcentral.com/articles/10.1186/s42238-022-00142-z ].
» https://jcannabisresearch.biomedcentral.com/articles/10.1186/s42238-022-00142-z - 2 Seddon T, Floodgate W. Regulating Cannabis [Internet]. Cham: Springer International Publishing; 2020. Available from: [http://link.springer.com/10.1007/978-3-030-52927-7].
» http://link.springer.com/10.1007/978-3-030-52927-7 - 3 Brasil. Ministério da Saúde. ANVISA. Relatório de Análise de Impacto Regulatório sobre Produtos de Cannabis para fins medicinais [Internet]. Brasília; 2024 [cited 2024 Oct 28]. Available from: [ https://www.gov.br/anvisa/pt-br/assuntos/regulamentacao/air/analises-de-impacto-regulatorio/2024/arquivos-relatorios-de-air-2024/relatorio-de-air-produtos-cannabis-medicinal-08082024.pdf ].
» https://www.gov.br/anvisa/pt-br/assuntos/regulamentacao/air/analises-de-impacto-regulatorio/2024/arquivos-relatorios-de-air-2024/relatorio-de-air-produtos-cannabis-medicinal-08082024.pdf - 4 Whiting PF, Wolff RF, Deshpande S, Di Nisio M, Duffy S, Hernandez AV, et al. Cannabinoids for medical use a systematic review and meta-analysis. JAMA [Internet]. 2015 Jun 23; 313(24): 2456-73. [cited 2022 Mar 23]. Available from: [ https://jamanetwork.com/ ].
» https://jamanetwork.com/ - 5 Bilbao A, Spanagel R. Medical cannabinoids: a pharmacology-based systematic review and meta-analysis for all relevant medical indications. BMC Medic. 2022 Aug 19; 20(1): 259. Available from: [https://pmc.ncbi.nlm.nih.gov/articles/PMC9389720/].
» https://pmc.ncbi.nlm.nih.gov/articles/PMC9389720/ - 6 Pinto CDBS, Esher A, Oliveira CVS, Osório-de-Castro CGS. Expansion of the medical cannabis market in Brazil and regulatory challenges. Cad Saúde Públ. 2024; 40(11): e00088624. [ https://doi.org/10.1590/0102-311XPT088624 ].
» https://doi.org/10.1590/0102-311XPT088624
-
Fontes de Financiamento:
Não houve financiamento ou suporte específico para a condução desse estudo.
Datas de Publicação
- Publicação nesta coleção
22 Ago 2025 - Data do Fascículo
Ago 2025
Histórico
- Recebido
20 Nov 2024 - Aceito
29 Jun 2025