Estado da Arte
Triterpenos como Inibidores de Fusão: Uma Nova Estratégia no Combate ao Vírus HIV
Triterpenes as Fusion Inhibitors: A New Strategy Against HIV Virus
Resumo
A Síndrome da Imunodeficiência Adquirida (SIDA) é uma das doenças mais assustadoras no mundo. Inúmeros esforços têm sido realizados na descoberta de novos agentes terapêuticos, capazes de combater o VIH-1 (Vírus da Imunodeficiência Humana tipo 1). Atualmente, a FDA (Food and Drug Administration) tem aprovado vinte e dois medicamentos anti-SIDA, classificados em quatro categorias; os nucleosídeo-nucleotídeo inibidores da transcriptase reversa (zidovudina, abacavir, lamivudina, estavudina, didadosina, zalcitabina, entricitabina e tenofovir), os não nucleosídeonucleotídeo inibidores da transcriptase reversa (nevirapina, efavirenz e delavirdina), inibidores de protease (saquinavir, ritonavir, lopinavir, indinavir, nelfinavir, amprenavir, tipranavir, fosamprenavir, darunavir e atazanavir) e os inibidores de fusão (enfurvitida). Esta última classe representa uma promissora área de pesquisa, pois previne a entrada do vírus VIH na célula hospedeira, sendo uma importante estratégia no combate ao surgimento de vírus resistentes aos medicamentos comumente utilizados. Devido a importância de estratégias alternativas no combate ao vírus VIH e a relevância dos produtos naturais na descoberta de novos fármacos, esse artigo tem como objetivo apresentar triterpenos antifusão como uma promissora classe de agentes anti-VIH.
- Unitermos:
- Triterpenos.
- inibidores de fusão.
- vírus VIH.
Abstract
Acquired immunodeficiency syndrome (AIDS) is one of the most frightening diseases worldwide. Extensive effort has been spent to discover therapeutic agents targeting the etiological agent of AIDS, human immunodeficiency virus type 1 (HIV-1). Currently, the FDA (Food and Drug Administration) has approved twenty two anti-AIDS drugs including nucleoside reverse transcriptase inhibitors (zidovudine, abacavir, lamivudine, stavudine, didadosine, zalcitabine, entricitabine and tenofovir), non-nucleoside reverse transcriptase inhibitors (nevirapine, efavirenz and delavirdine), protease inhibitors (saquinavir, ritonavir, lopinavir, indinavir, nelfinavir, amprenavir, tipranavir, fosamprenavir, darunavir and atazanavir) as well fusion inhibitors (enfurvitide). This last category represents a promising strategy, due to it prevents the entrance of virus HIV in the target cell, being an important weapon in the combat to the appearance of resistant virus to medicines used. Due to the importance of alternative strategies in the fight to virus HIV and the relevance of the natural products in the discovery of new drugs, the aim of this article is to highlights triterpenes antifusing as a promising category of anti-HIV agents.
- Key Words:
- Triterpenes.
- fusion inhibitors.
- virus HIV.
Introdução
A Síndrome da Imunodeficiência Adquirida (SIDA) ou Acquired Immuno Deficiency Syndrome (AIDS) é um alarmante problema de saúde pública mundial. Por exemplo, estima-se que, em 2005, 38,6 milhões de pessoas eram portadoras do vírus VIH (Vírus da Imunodeficiência Humana) ou HIV (Human Immunodeficiency Virus), sendo esse responsável por cerca de 2,8 milhões de óbitos. Esse quadro é ainda mais grave em países subdesenvolvidos, principalmente na África SubSahariana, onde até o final de 2005, existiam 24,5 milhões de pessoas vivendo com HIV, ou seja, 64% do número total de casos no mundo estão concentrados nessa região (UNESCO, 2006). O espectro dessa doença já atingiu uma dimensão cujos reflexos começam a atingir o desenvolvimento econômico e social das nações, estando presente em mais de 190 países, com aproximadamente 95% dos infectados presentes em países subdesenvolvidos (WHO, 2006).
O agente etiológico da AIDS é o HIV, um retrovírus que necessita instalar-se no interior de células, utilizando-se do material genético das mesmas para se reproduzir. Seu alvo são os linfócitos T do tipo CD4+ que integram o nosso sistema imunológico. Ao atingir estas células, o vírus leva a uma descoordenação desse sistema, provocando uma grave imunodeficiência, por meio da qual o indivíduo portador torna-se mais susceptível a infecções por agentes oportunistas, como a tuberculose (TB), por exemplo (DE SOUZA; DE ALMEIDA, 2003). Estima-se que, em todo o mundo, um terço dos quase 40 milhões de pessoas que vivem com HIV/AIDS estejam co-infectados com TB, sendo 70% desses casos, relatados em países da África SubSahariana (WHO, 2006). Sabe-se que o risco de progressão da infecção com Mycobacterium tuberculosis, até o desenvolvimento da doença aumenta de acordo com a imunodepressão do indivíduo. Sendo assim, pessoas infectadas com HIV têm esse risco dez vezes superior, se comparados com indivíduos sadios (HARRIES et al., 2004).
O tratamento da AIDS baseia-se na terapia antiretroviral combinada (HAART) e, desde que foi aprovada em 1996, essa terapia trouxe consideráveis benefícios aos portadores da doença, proporcionandolhes um maior tempo de sobrevida, reduzindo à freqüência de internações ocasionadas devido a infecções oportunistas, permitindo que os pacientes tenham uma melhor qualidade de vida. Atualmente, existem 22 medicamentos aprovados pela FDA no combate ao HIV que atuam inibindo etapas específicas do ciclo viral, suprimindo a replicação do vírus, o que impede a infecção de novas células (FDA, 2006). Estes se dividem em quatro classes: os nucleosídeos inibidores da transcriptase reversa (zidovudina, abacavir, lamivudina, estavudina, didadosina, zalcitabina, entricitabina e tenofovir) que impedem a replicação do vírus incorporando-se à cadeia de DNA que será produzida pela ação da enzima viral transcriptase reversa; os não nucleósidos inibidores da transcriptase reversa (nevirapina, efavirenz e delavirdina) que se ligam a transcriptase reversa, bloqueando seu funcionamento e impedindo a produção do DNA viral; os inibidores de protease (saquinavir, ritonavir, lopinavir, indinavir, nelfinavir, amprenavir, tipranavir, fosamprenavir, darunavir e atazanavir) que atuam impedindo que novos vírus amadureçam e infectem outras células e os inibidores de fusão (enfuvirtida). (PEÇANHA et al., 2002).
Inibidores de fusão
Trata-se de uma nova classe de fármacos antiretrovirais muito promissora, visto que são capazes de impedir a infecção de novas células, por não permitirem a entrada do vírus nas mesmas (DE SOUZA, 2004; GALLO et al., 2003). Para que se entenda como agem esses medicamentos, é necessário compreender os mecanismos moleculares envolvidos no processo de fusão do vírus à célula hospedeira (FIGURA 1). Ao entrar no organismo humano e chegar à corrente sanguínea, o vírus se encontra com os linfócitos auxiliares do tipo T, macrófagos e outras células do sistema imunológico, que expressam em sua superfície uma glicoproteína denominada CD4. A glicoproteína externa gp120, presente na membrana viral, interage com o CD4, contando com o auxílio de co-receptores, que são moléculas de adesão de origem celular, denominadas CCR5 e/ou CXCR4, que promovem a ligação entre o vírus e a célula-alvo. Uma vez concretizada esta ligação, a glicoproteína transmembranar viral gp41 sofre uma alteração conformacional que promove a exposição de um domínio hidrófobo, denominado peptídeo de fusão, permitindo assim a fusão do vírus à membrana da célula hospedeira (DE SOUZA, 2004).
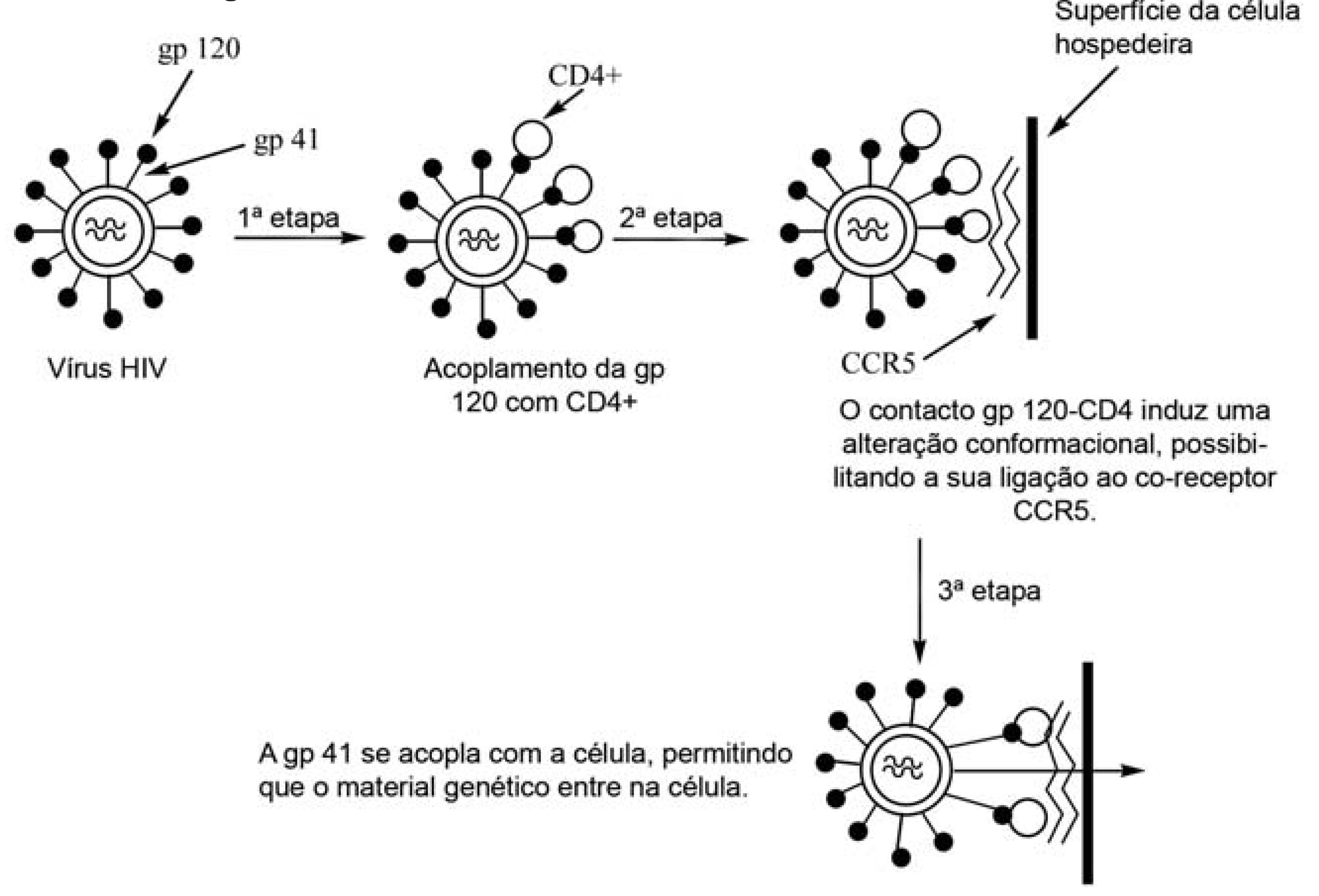
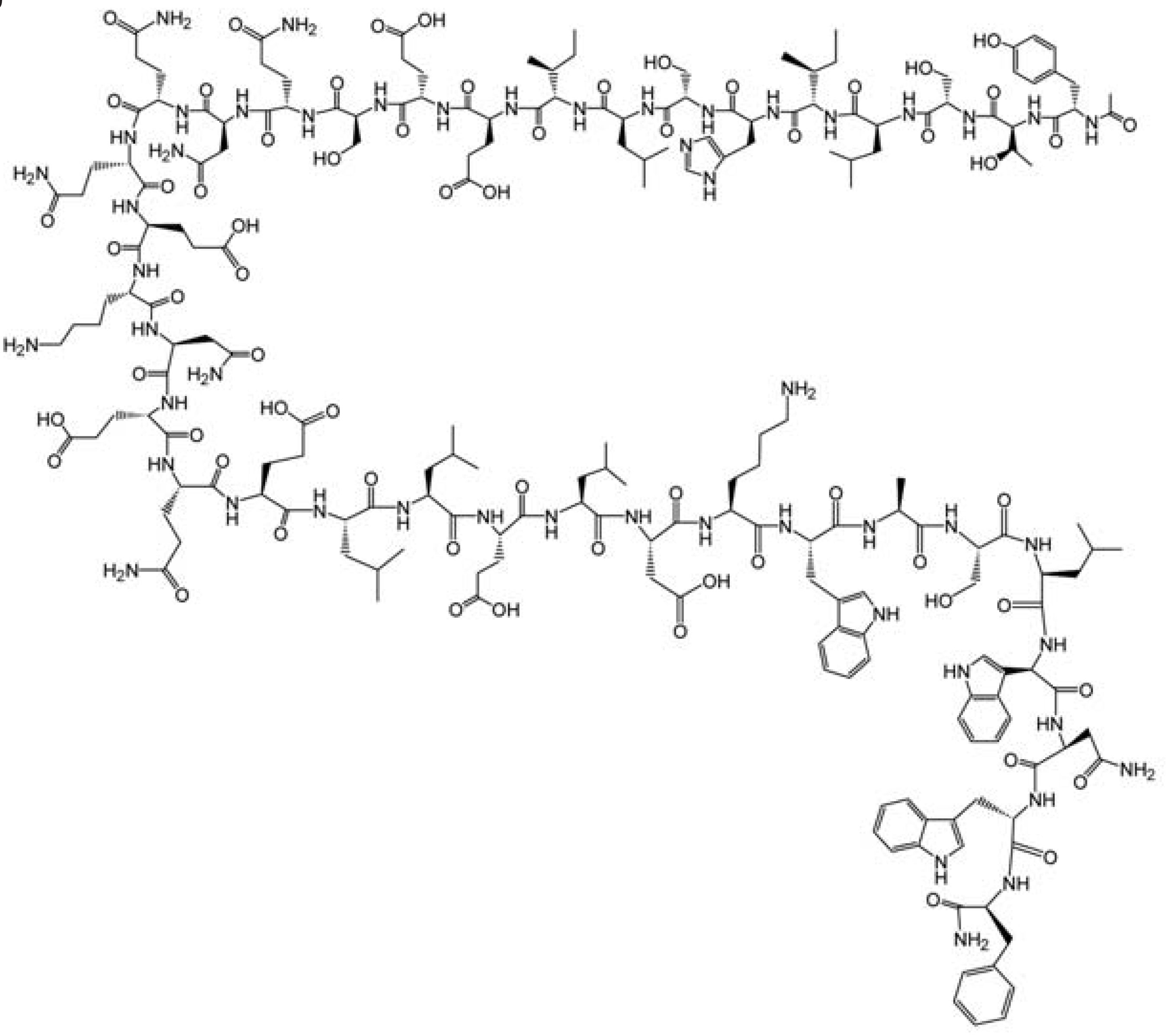
O único fármaco desta classe existente no mercado é o Fuseon® , também conhecido como T-20, enfuvirtida e pentafusida, que foi desenvolvido e fabricado pela Roche Holding AG (Suíça) e Trimeris Inc. (USA), sendo aprovado pela FDA (Food and Drug Administration) em março de 2003 e em maio desse mesmo ano pela Comissão Européia (HUNTER et al., 2000). Este é um peptídeo com 36 resíduos de aminoácidos de seqüência idêntica a da alfa-hélice mais próxima do terminal carboxílico e da região transmembranar da gp41, denominada HR2, cuja seqüência pode ser representada por: CH3 CO-Tir-Tre-Ser-Leu-Ile-His-Ser-Leu-Ile-Glu-Glu-Ser-Gln-Asn-Gln-Gln-Glu-Lis-Asn-Glu-Gln-Glu-Leu-Leu-Glu-Leu-Asp-Lis-Trp-Ala-Ser-Leu-Trp-Asn-Trp-Fenil-NH2 (FIGURA 2) (DE SOUZA, 2005). Sendo assim, quando uma cópia dessa região é introduzida no organismo, essa se liga de modo competitivo a HR1, impedindo que a gp41 se dobre, o que promove a inibição da fusão entre o vírus e a célula-alvo (DE SOUZA, 2005).
Para se ter idéia de quanto este medicamento é inovador e promissor, um estudo realizado com 70 pessoas durante 48 semanas, demonstrou que 56% dos pacientes tratados com 50mg de Fuzeon® por injeção subcutânea duas vezes ao dia, em associação à terapia oral convencional otimizada por testes de resistência genotípica, apresentaram melhorias significativas, com um decréscimo para menos de 400 cópias de RNA de HIV-1 por mililitro de sangue. Estes resultados permitem concluir que regimes terapêuticos de combinação do Fuseon® com outros compostos antiretrovirais são eficazes para ultrapassar resistências adquiridas (RIMSKY et al., 1998). Além disso, um estudo envolvendo o acompanhamento de 39 doentes durante 3 anos demonstrou que a resistência primária ao tratamento com T-20 é um acontecimento raro (ZÖLLNER et al., 2001).
Mesmo com todos os avanços apresentados pela terapia anti-retroviral atualmente utilizada, existem alguns problemas relacionados ao aparecimento de cepas resistentes do vírus. Por exemplo, no Brasil, em pesquisa realizada com 500 pacientes da Rede Nacional de Genotipagem (Renageno) demonstrou que somente 7% não apresentavam resistência a nenhum dos antiretrovirais. Nos Estados Unidos, em 1998, a resistência primária a esses fármacos era igual a 3,5%, já em 2000, esta passou para 14%. A resistência pode ser conseqüência ou causa da falha terapêutica. No primeiro caso, o anti-retroviral deixa de atuar devido à falta de adesão do paciente ao tratamento, já no outro a resistência acontece devido a mutações sofridas pelo vírus, dada a freqüência e a constância de contato com os fármacos (SBI, 2003). Tendo em vista a evolução da epidemia mundial da AIDS, bem como os diversos inconvenientes relacionados à terapia anti-retroviral atualmente empregada, faz-se necessário o desenvolvimento de novos fármacos no combate ao vírus HIV.
Produtos naturais no combate ao vírus HIV
A pesquisa de produtos naturais é uma importante ferramenta no desenvolvimento de novos padrões moleculares bioativos, servindo como modelo para criação de novos análogos sintéticos ou semi-sintéticos. Para se ter uma idéia de quanto os produtos naturais são importantes para o desenvolvimento de novos fármacos, sabe-se que das 520 novas drogas aprovadas no período de 1983 a 1994, 39% são produtos naturais ou derivados e, ainda, que 60-80% das drogas antibacterianas e anticancerígenas também são derivadas desses (CRAGG et al., 1997). Outro dado interessante é que dos 20 medicamentos mais vendidos em 1999, oito (simvastatina, lovastatina, enalapril, pravastatina, atorvastatina, augmentina, claritromicina e ciclosporina) derivam de produtos naturais, movimentando mais de 16 bilhões de dólares nesse mesmo ano (HARVEY, 2000).
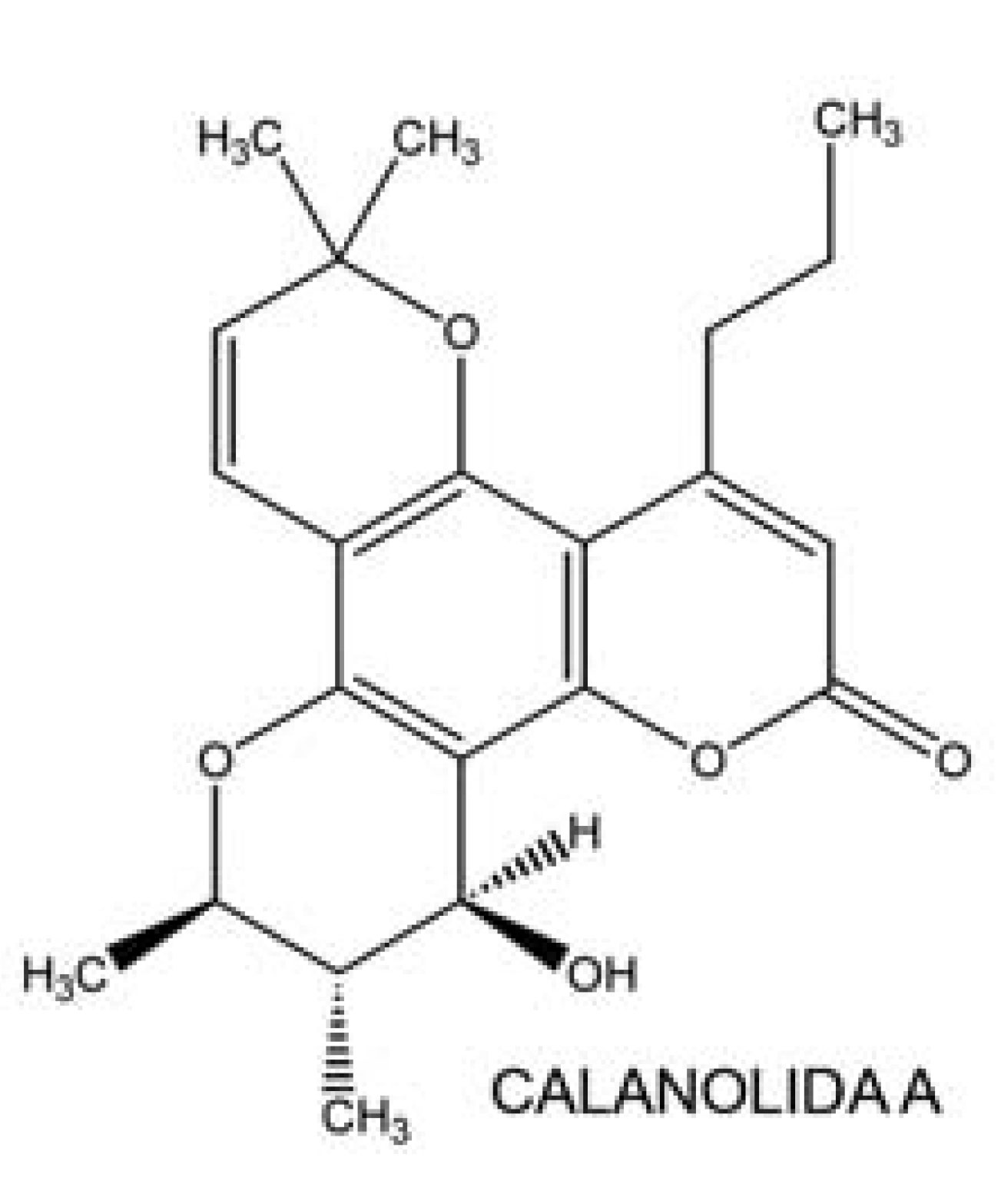
Atualmente, existem relatados na literatura inúmeros estudos mostrando produtos naturais com atividade antiretroviral, tendo como origem as mais diversas fontes (SINGH et al., 2005), como plantas, fungos, bactérias e organismos marinhos (JUNG et al., 2000). Esses produtos têm demonstrado um grande potencial principalmente no desenvolvimento de novos quimioterápicos, devido sua baixa toxicidade e sua atividade específica. Dentre as diversas moléculas promissoras existentes, destaca-se a Calanolida A (FIGURA 3), uma cumarina extraída a partir de várias espécies de plantas do gênero Callophy llum. Esta se encontra em fase II de testes clínicos, enquadrando-se no grupo dos nãonucleosídeos inibidores da transcriptase reversa.
A partir da necessidade do desenvolvimento de novas terapias contra o HIV e tendo em vista a importância dos produtos naturais para tal, o objetivo dessa revisão é divulgar o estudo de triterpenos como possíveis inibidores de fusão.
Triterpenos como agentes antifusão
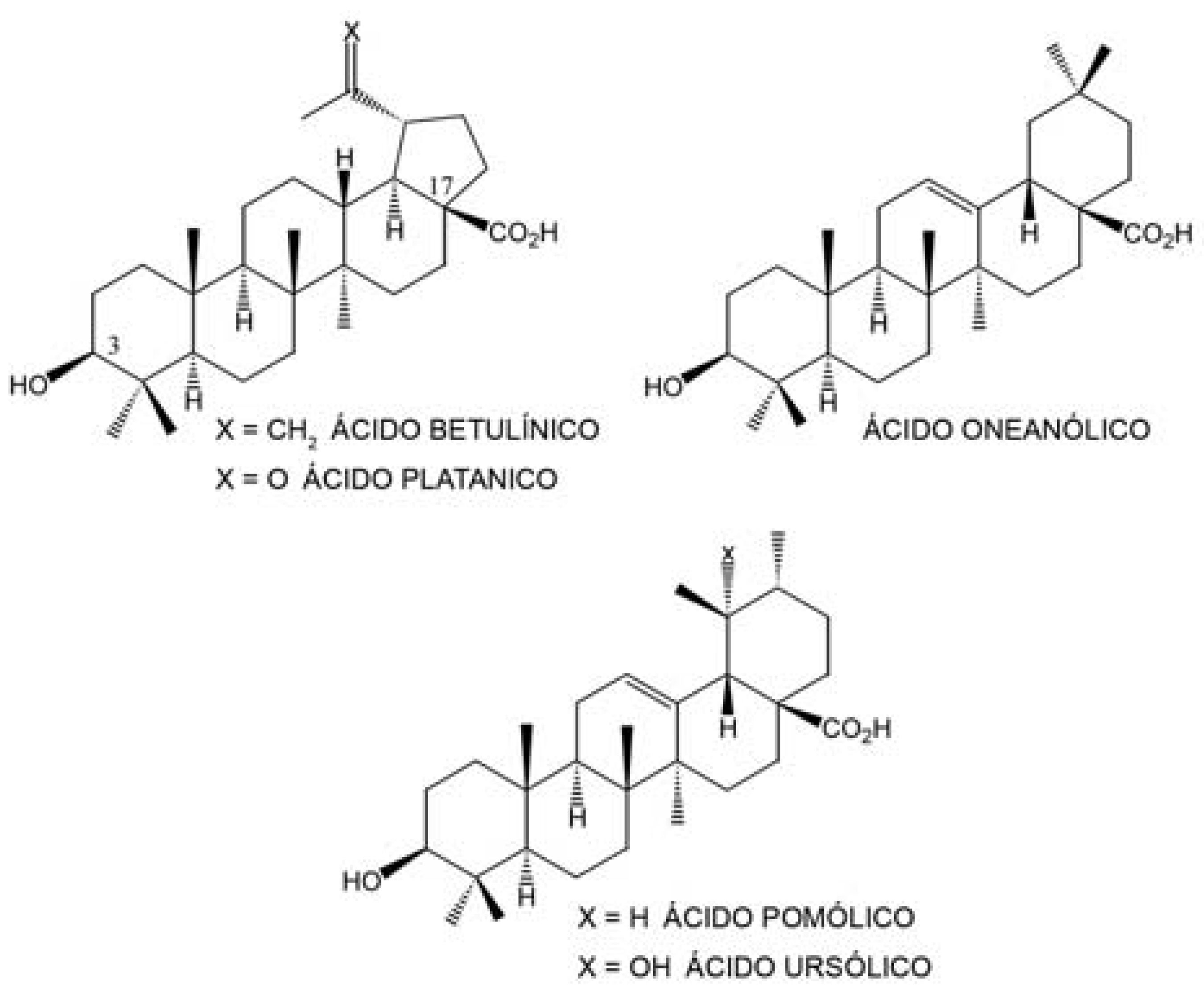
Os triterpenos constituem uma promissora classe de compostos anti-HIV (SUN et al., 2003) que, devido a seus promissores resultados no combate à replicação viral, vem recebendo especial atenção da comunidade científica (HUANG, 2002). Esses referidos compostos são capazes de atuar em diferentes etapas do ciclo de replicação viral (SUN et al, 2002), como na maturação (KANAMOTO et al., 2001), na transcriptase reversa (SUN et al, 2003) e na fusão entre o vírus e os linfócitos (HOLZ-SMITH et al., 2001). Os primeiros triterpenos a apresentarem atividades anti-HIV foram os ácidos betulínico e platânico (FIGURA 4), que ocorrem em Syzigium claviflorum (Myrtaceae), exibindo atividade inibitória contra a replicação do vírus HIV-1 em células linfocitárias H9 com valores de EC50 (atividade anti-HIV) de 1.4 e 6.5 μM e valores de TI (Índice Terapêutico) de 9,3 e 14, respectivamente (FUJIOKA et al., 1994). Posteriormente, outros triterpenos anti-HIV foram identificados, como o ácido oleanólico (FIGURA 4), EC50 = 3.7 μM e TI 12,8, isolado de diferentes plantas, como a Rosa woodsii (Rosaceae), Prosopis glandulosa (Leguminosae), Phoradendron juniperinum (Loranthaceae), Syzigium claviflorum (Mirtaceae), Hyptis capitata (Lamiaceae) e Ternstromia gymnanthera (Theacea) (KASHIWADA et al., 1998). Dois outros triterpenos apresentam também atividade anti-HIV, o ácido pomólico, EC50 = 3.0 μM e TI 16,3 e o ácido ursólico, EC50 = 4,4 μM e TI 3,3 (FIGURA 4), os quais são isolados a partir da Prosopis glandulosa, Phoradendron juniperinum, Syzigium claviflorum e Hyptis capitata (KASHIWADA et al., 2000).
Com o objetivo de se obter novos fármacos, diferentes análogos foram sintetizados e avaliados. Estes análogos normalmente são realizados via hemisíntese a partir do triterpeno, sendo as modificações comumente realizadas na hidroxila em posição C-3 e no ácido carboxílico em posição C-17 (FIGURA 5). Um importante triterpeno hemisintético, conhecido como RPR 10361 (FIGURA 5), é um composto de baixa massa molar não peptídico capaz de interagir com a gp 41 (LABROSSE et al., 2000), bem como estabilizar o complexo gp120-gp41 (LABROSSE et al., 1997), bloqueando assim a infecção viral em concentrações inferiores a 10 nM. Seu estereoisômero, o IC9564 (FIGURA 5), é também um potente inibidor de fusão, capaz de atuar na gp120 viral (HOLZ-SMITH et al., 2001).
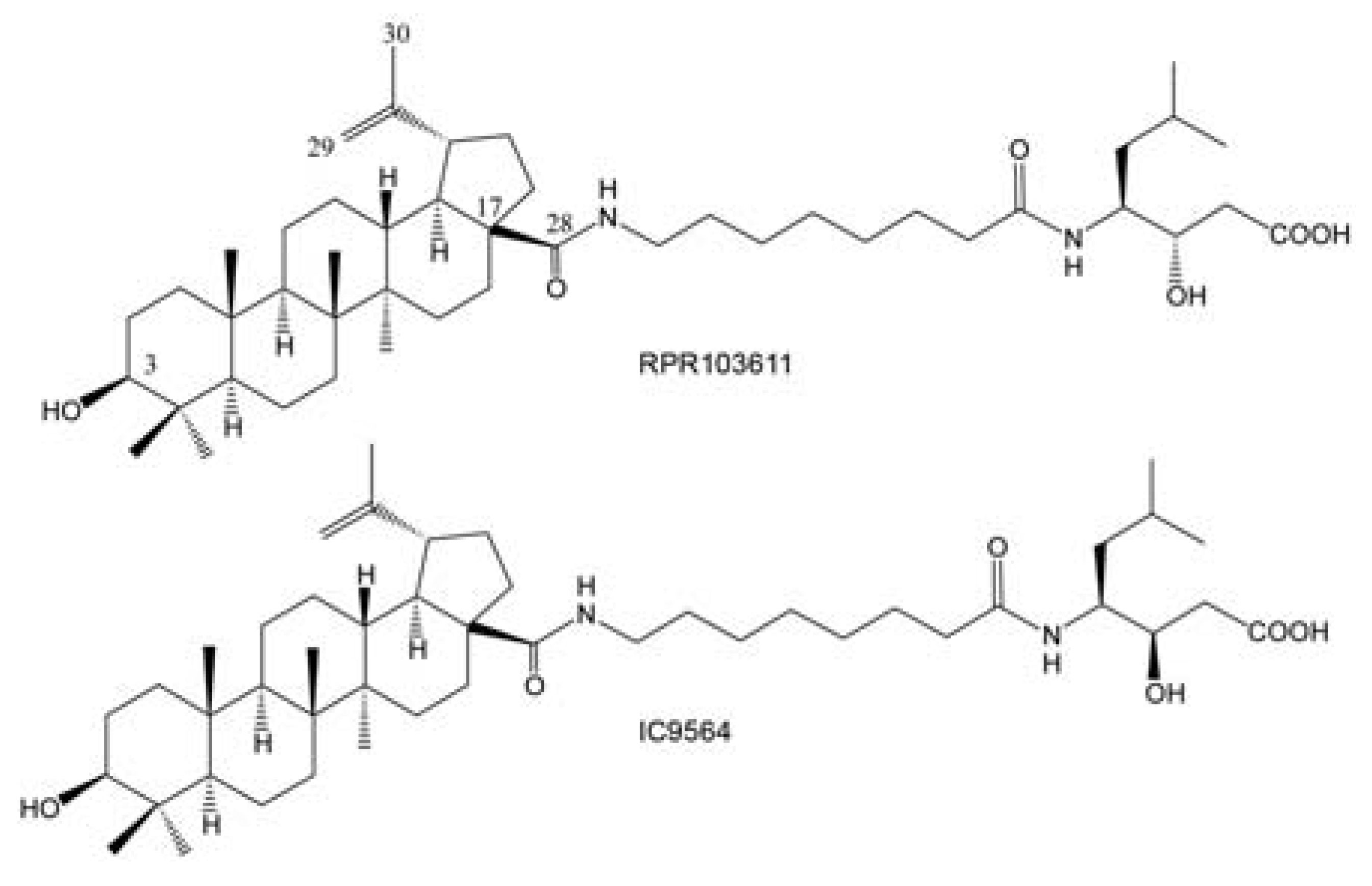
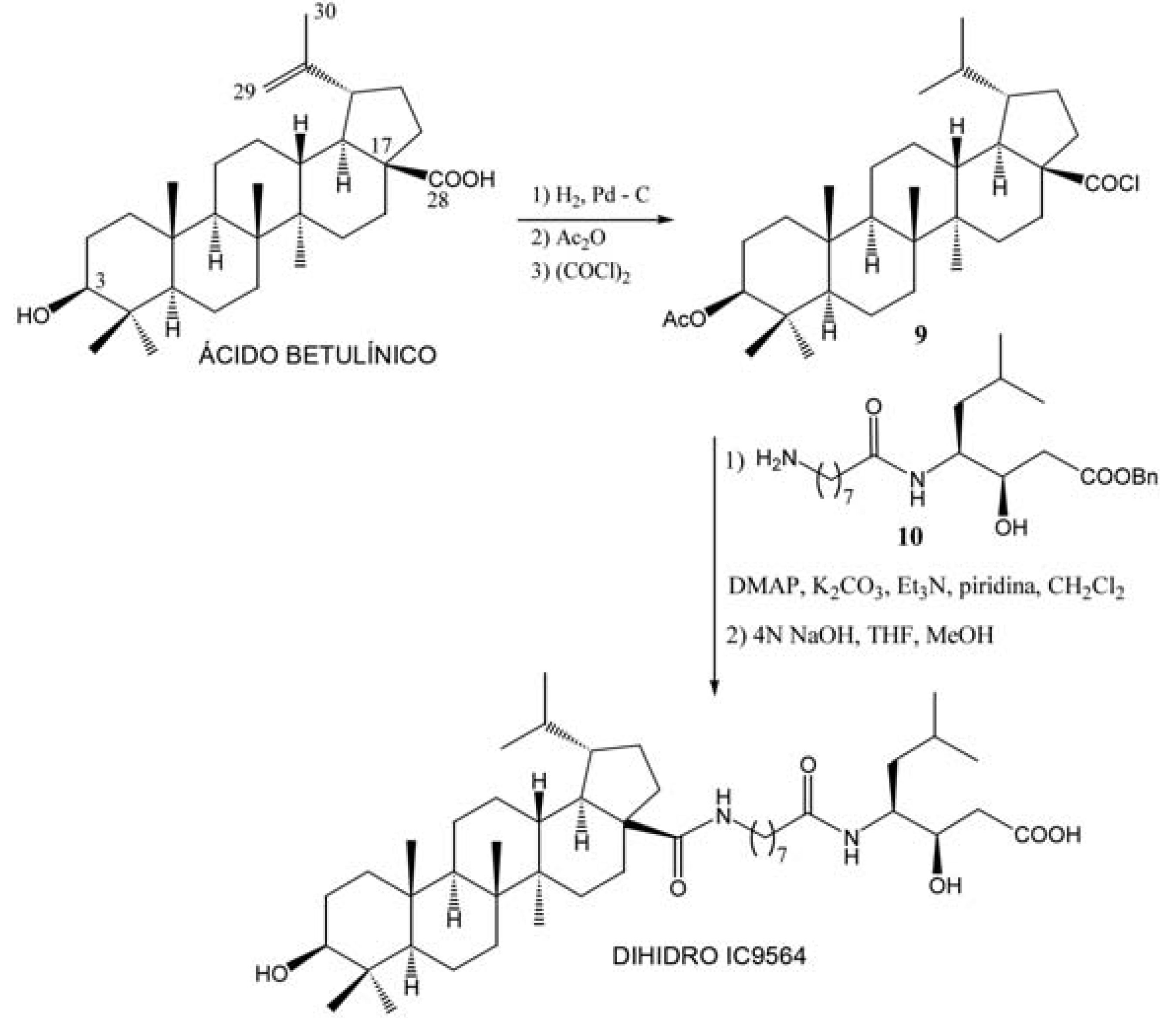
Devido aos bons resultados do RPR103611 e IC9564 (FIGURA 5), análogos deste composto têm sido sintetizados. Como exemplo, podemos citar a obtenção do dihidro IC9564 (FIGURA 6), que é preparado via hemisíntese a partir do ácido betulínico (SUN et al., 2003) que, após hidrogenação da dupla ligação, acetilação da hidroxila em posição C-3, em presença de anidrido acético e formação do cloreto de acila em posição C-28, utilizando-se cloreto de oxalila, fornece o intermediário 9. O acoplamento sob condições básicas entre os intermediários 9 e 10 (HOLZ-SMITH et al., 2001), seguido da desproteção dos grupos acetila e benzila em presença de hidróxido de sódio, fornece a molécula alvo, dihidro IC9564 (FIGURA 6). A avaliação antifusão do composto dihidro IC9564, EC50 = 0,33 μM, em células infectadas H9, mostrou ser equipotente ao IC9564, EC50 = 0,4 μM, indicando que a dupla ligação do grupo isopropilideno poderia não ser importante para a atividade biológica. No entanto, o seu intermediário precursor protegido apresenta um EC50 > 28 μM, o que indica a importância da hidroxila (C-3) e do ácido carboxílico (C-28) livre para a atividade biológica.
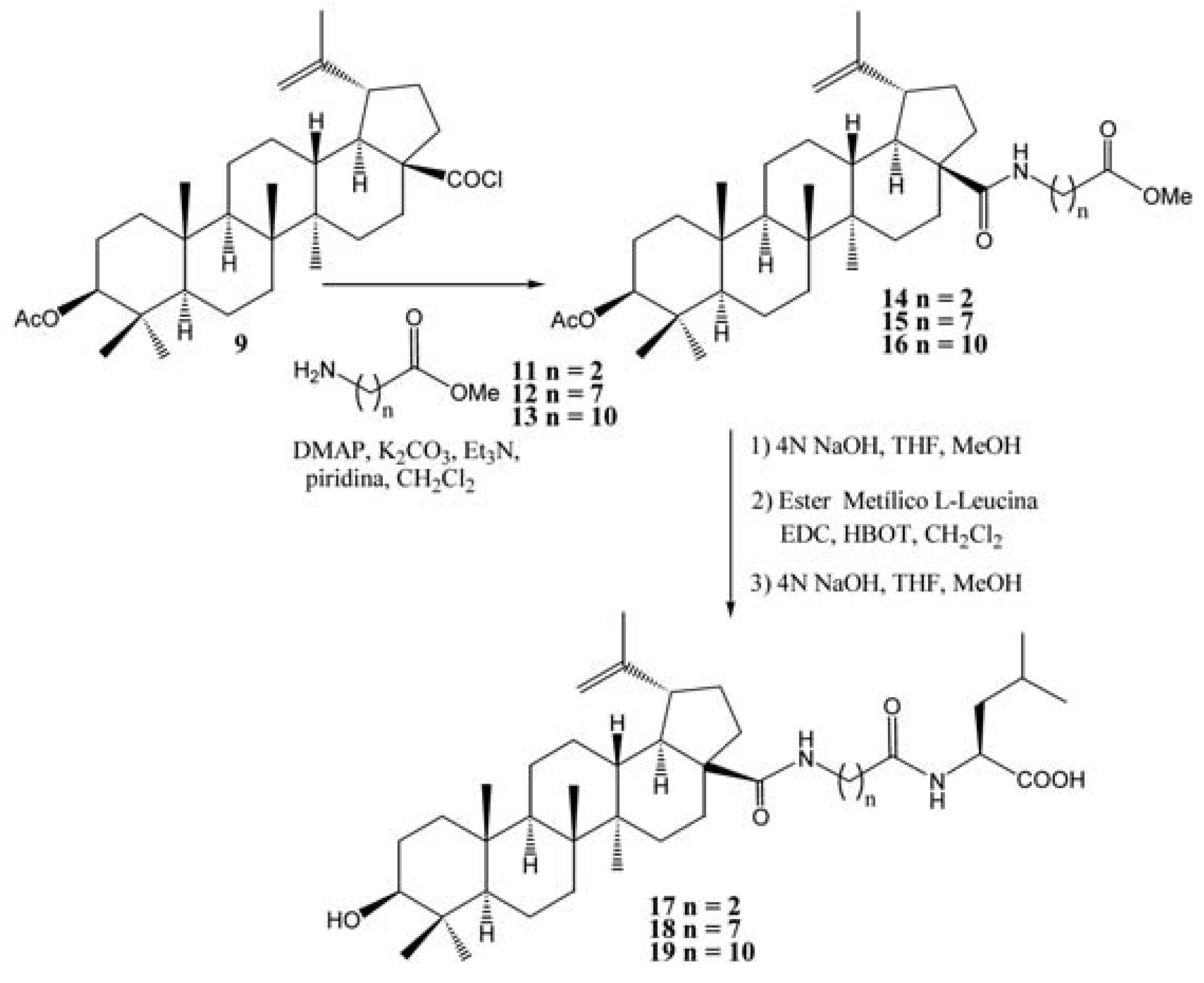
Um outro exemplo de análogos do RPR103611 e IC9564 (FIGURA 5) são os compostos 17-19 (FIGURA 7) (SUN et al., 2002), obtidos via hemisíntese a partir do ácido betulínico e de maneira similar ao descrito no esquema da FIGURA 6. Após o acoplamento em condições básicas entre o triterpeno 9 e os fragmentos 11, 12, e 13, foram obtidos os intermediários 14-16 (FIGURA 7), que seguido da saponificação do éster em presença de hidróxido de sódio, acoplamento peptídico e finalmente, desproteção do éster em presença de hidróxido de sódio produz os análogos 17-19 (Esquema 2). Estes análogos foram avaliados quanto a sua atividade antifusão em células infectadas H9, com o composto 17, apresentando perda de atividade EC50 > 28 μM em relação aos compostos RPR103611 (0,33 μM) e IC9564 (0,4 μM) (FIGURA 5). No entanto, os compostos 18 e 19 apresentaram um aumento na atividade antifusão com os EC50 de 1,1 e 0,33 μM, respectivamente, indicando a importância do número de átomos de carbono da cadeia lateral.
Além de sua promissora atividade como agentes inibidores de fusão, os derivados do ácido betulínico também podem atuar em diferentes etapas do ciclo de replicação do HIV. Como exemplo, podemos citar o PA-457 (Bervirimat) (FIGURA 8), que pode ser considerado o primeiro fármaco de uma nova classe de medicamentos contra o vírus HIV: os inibidores de maturação, capaz de inibir o último estágio do ciclo reprodutivo do vírus HIV (LI et al., 2003). Esse produto natural vem sendo desenvolvido pela Panacos Pharmaceuticals, e se encontra em fase II de testes clínicos (COHLAN, 2006).
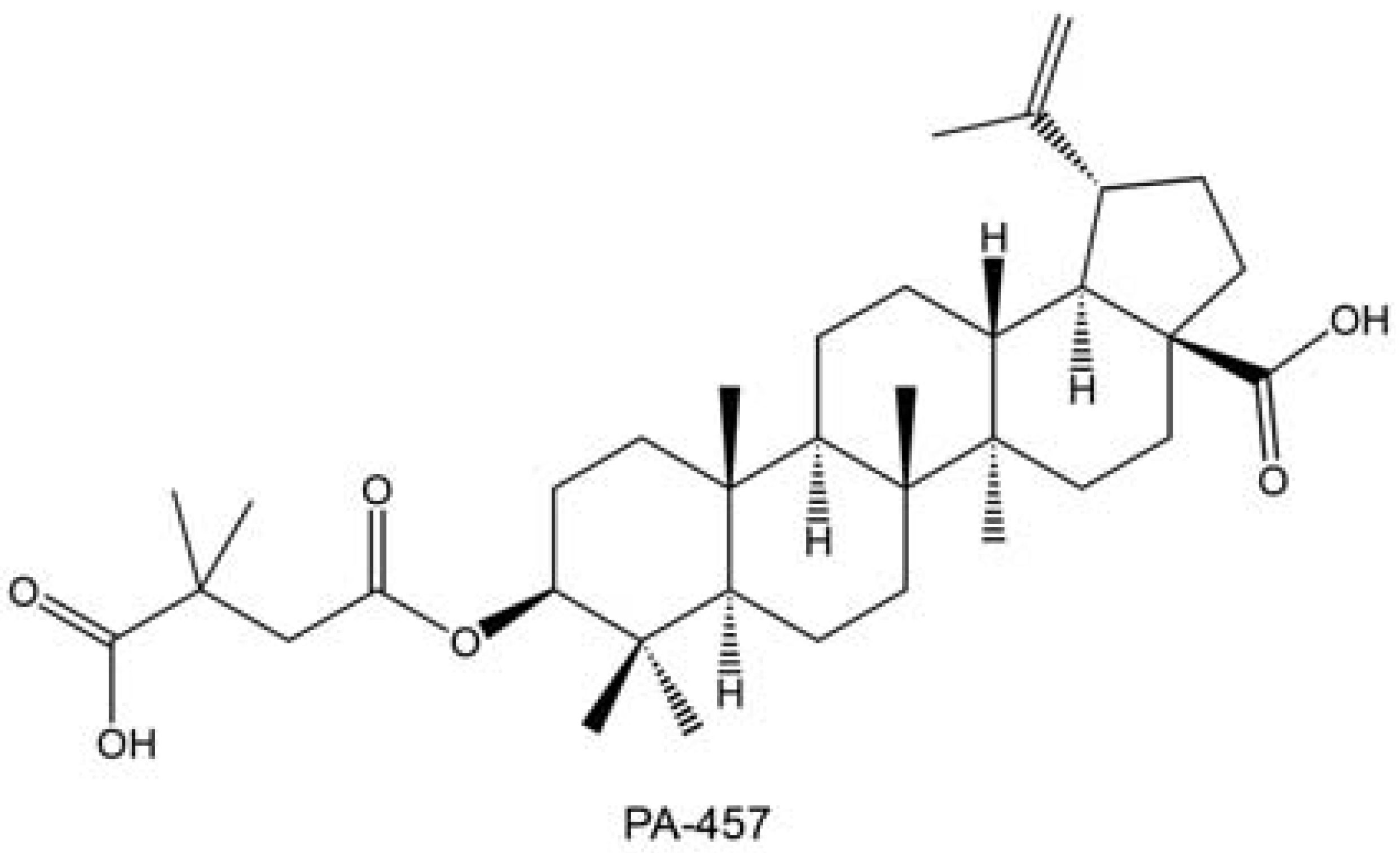
Um ensaio preliminar em humanos, relatado em agosto de 2005, mostrou que o PA-457 reduziu em 10 vezes o HIV presente no sangue, em apenas algumas horas. Em breve, também será ensaiado em combinação com outros fármacos contra o HIV e testado, num ensaio controlado por placebo, em 48 pacientes cujos tratamentos estão em falência. No entanto, estima-se que esse fármaco esteja disponível no mercado somente em 2009 (COHLAN, 2006). Tendo em vista esse contexto, pode-se concluir que triterpenos naturais e sintéticos são uma classe de compostos capazes de atuarem como inibidores de fusão, representando uma moderna estratégia no combate à replicação viral, impedindo que o vírus infecte as células e nem sequer inicie a infecção destas. Essa estratégia vem apresentando promissores resultados, recebendo destaque como uma nova geração de fármacos anti-HIV.
Referências
COHLAN, A. Trials for drug that leaves HIV defenceless. New Scientist Magazine, v.2555, n.16, 2006.
DE SOUZA, M. V. N.; DE ALMEIDA, M. V. Drogas anti-HIV: Passado, presente e perspectivas futuras. Química Nova, v.26, n.3, p.366-372, 2003.
DE SOUZA, M.V.N. Entry Inhibitors - A New Class of Aids Drugs. Letters in Drug Design & Discovery, v.1, p.184-193, 2004.
DE SOUZA, M.V.N. Fármacos Inibidores de Fusão: uma Nova Estratégia no Combate à Replicação do Vírus VIH. Acta Farmacéutica Bonaerense, v.24, n.2, p.291-9, 2005.
DE SOUZA, M.V.N. Fuzeon, o primeiro medicamento de uma nova classe anti-HIV denominada inibidores de fusão. Revista Brasileira de Farmácia, v.86, n.3, p.112-116, 2005.
CRAGG, G.M.; NEWMAN, D.J.; SNADER; K.M. Natural Products in Drug Discovery and Development. The Journal of Natural Products, v.60, p.52-60, 1997.
FDA: FOOD AND DRUG ADMINISTRATION. http://www.fda.gov/oashi/aids/hiv.html; Accessed in 19/12/2006.
FUJIOKA, T.; KASHIWADA, Y.; KIKUSKIE, R. E.; COSENTINO, L. M.; BALLAS, L. M.; JIANG, J. B.; JANZEN, W. P.; CHEN, I. S.; LEE, K. H. The Journal of Natural Products, v.57, p.243-247, 1994.
GALLO, S.A.; FINNEGAN, C.M.; VIARD, M.; RAVIV, Y.; DIMITROV, A.; RAWAT, S.S.; PURI, A.;DURELL, S.; BLUMENTHAL, R. The HIV Env-mediated fusion reaction. Biochimica et Biophysica Acta, v.1614, p.36-50, 2003.
HARRIES, A. D., MAHER, D., GRAHAM, S. TB/HIV: Manual Clínico, 2a ed., WHO/HTM/TB/2004.329
HARVEY, A. Strategies for discovering drugs from previously unexplored natural products. Drug Discovery Today, v.5, n.7, p. 294-300, 2000.
HOLZ-SMITH, S.L.; SUN, I.C.; JIN, L.; MATTHEWS, T.J.; LEE, K.H.; CHEN, C.H. Role of human immunodeficiency virus (HIV) type 1 envelope in the anti-HIV activity of the betulinic acid derivative IC9564. Antimicrobial Agents and Chemotherapy, v. 45, n.1, p.60-66, 2001.
HUANG, L.; CHEN, C. H. Molecular targets of anti-HIV-1 triterpenes. Current Drugs Targets Infection Disorders v.2, n.1, p.33-36, 2002.
HUNTER, E. et. al. Sensitivity of Human Immunodeficiency Virus Type 1 to the Fusion Inhibitor T-20 Is Modulated by Coreceptor Specificity Defined by the V3 Loop of gp120. Journal of Virology, v.74, n.18, p.8358-8367, 2000.
JUNG, M., KIM, H., LEE, S., KIM, H. Recent Studies on Natural Products as Anti-HIV Agents. Current Medicinal Chemistry, v.7, p.649-661, 2000
KANAMOTO, T.; KASHIWADA, Y.; KANBARA, K.; GOTOH, K.; YOSHIMORI, M.; GOTO, T.; SANO, K.; NAKASHIMA, H. Anti-human immunodeficiency virus activity of YK-FH312 (a betulinic acid derivative), a novel compound blocking viral maturation. Antimicrobial Agents and Chemotherapy, v. 45, n.4, p.1225-1230, 2001.
KASHIWADA, Y.; WANG, H., K.; NAGAO, T.; KITANAKA, S.; YASUDA, I.; FUJIOKA, T.; YAMAGISHI, T.; COSENTINO, M.; KOZUKA, M.; OKABE, H.; IKESHIRO, Y.; HU, C. Q.; YEH, E.; LEE, K. H. Anti-AIDS Journal of Natural Products, v.61, p.1090-1095, 1998.
KASHIWADA, Y.; NAGAO, T.; HASHIMOTO, A.; IKESHIRO, Y.; OKABE, H.; COSENTINO, L. M.; LEE, K. H. Anti-AIDS Agents 38. Anti-HIV activity of 3- O-acyl ursolic acid derivatives. The Journal of Natural Products, v.63, p.1619-1622, 2000.
LABROSSE, B.; PLESKOFF, O.; SOL, N.; JONES, C.; HENIN, Y.; ALIZON, M. Resistance to a drug blocking human immunodeficiency virus type 1 entry (RPR103611) is conferred by mutations in gp41. Journal of Virology, v.71, n.11, p.8230-8236, 1997.
LABROSSE, B.; TREBOUTE, C.; ALIZON, M. sensitivity to a non-pepitidic compound (RPR103611) blocking human immunodeficiency virus type env-mediated fusion depends on sequence and accessibility of the gp41 loop region. Journal of Virology, v.74, p.2142-2150, 2000.
LI, F.; GOILA-GAUR, R.; SALZWEDEL, K.; KILGORE, N.R.; REDDICK, M.; MATALLANA, C.; CASTILLO, A.; ZOUMPLIS, D.; MARTIN, D.E.; ORENSTEIN, J.M.; ALLAWAY, G.P.; FREED, E.O.; WILD, C.T. PA-457: A potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proceedings of the National Academy of Sciences, v.100, n.23, p.13555-13560, 2003.
PEÇANHA, E.P., ANTUNES, O.A.C., TANURI, A. Estratégias farmacológicas para a terapia anti-AIDS. Quimica Nova, v. 25, n. 6B, p.1108-1116, 2002.
RIMSKY, L.T.; SHUGARS, D.C.; MATHEWS, L.J. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides, Journal of Virology, v.72, p.986-993, 1998.
SINGH, I.P., BHARATE, S.B., BHUTANI, K.K.. Anti-HIV natural products. Current Science, v.89, n.2, p. 269-290, 2005.
SBI: SOCIEDADE BRASILEIRA DE INFECTOLOGIA. Tratamento Hoje: Boletim Terapêutico de HIV/Aids, DST e Hepatites virais, ano I, v.3, p. 1-4, 2003.
SUN, I. C.; CHEN, C. H.; KASHIWADA, Y.; WU, J. H.; WANG, H. K.; LEE, K. H. Anti-AIDS agents 49. Synthesis, anti-HIV, and anti-fusion activities of IC9564 analogues based on betulinic acid. Journal of Medicinal Chemistry, v.45, p.4271-4275, 2002.
SUN, I. C.; KASHIWADA, Y.; MORRIS-NATSCHKE, S.L.; LEE, K.H. Plant-derived terpenoids and analogues as anti-HIV agents. Current Topical in Medicinal Chemistry, v.3, n.2, p.55-169, 2003.
UNESCO. Report on the global AIDS epidemic, 2006. www.fda.gov/oashi/aids/virals.html. Acesso em 18/10/06; www.who.int/hiv/en/. Acesso em 20/10/06; www.who.int/tb/hiv/faq/en/index.html. Acesso em 17/10/06.
WHO: WORLD HEALTH ORGANIZATION. http://www.who.int/topics/hiv_infections/en/; Accessed in 19/12/2006.
ZÖLLNER, B.; FEUCHAT, H.-H.; SCHRÖTER, M.; SCHÄFER, P.; PLETTENBERG, A.; STOEHR, A.; LAUFS, R. Primary genotypic resistance of HIV-1 to the fusion inhibitor T-20 in long-term infected patients. AIDS, v.15, p.935-936, 2001.