Estado da Arte
Sobre a Legislação de Registro de Fitoterápicos1
On the Legal Requirements for the Registration of Phytomedicines
Resumo
O aprimoramento do conhecimento científico e tecnológico referente aos fitoterápicos; paralela e indiretamente, tem ocasionado uma evolução da legislação para o registro de tais produtos, pelas autoridades de saúde. O presente artigo traz uma revisão dos processos legais para o registro de fitoterápico no Brasil, assim como de alguns casos de legislações estrangeiras sobre o tema.
- Unitermos:
- Legislação.
- Fitoterápicos.
- Registro.
Abstract
Continuous and constant scientific and technological advances concerning phytomedicines, has led to a natural evolution on the legal procedures for the registration of such products, by the health authorities. The present article reviews the evolution of registration procedures for phytomedicines in Brazil as well as brings a brief description on how some foreign countries deal with this matter.
- Key words:
- Regulations.
- Phytomedicines.
- Application.
Introdução
A regulação e o controle de produtos disponíveis para venda no mercado é uma atividade de vigilância sanitária e uma prerrogativa do Estado. A primeira forma de controle é o registro do produto na Agência Nacional de Vigilância Sanitária - Anvisa, a qual verificará se aquele cumpre os requisitos que permitam a sua comercialização. Estes requisitos são definidos por intermédio do processo de consulta pública, e posteriormente são publicados na forma de Resoluções pela Anvisa. No caso específico de medicamentos, podem-se dividir os requisitos em dois grandes grupos: os requisitos tecnológicos, que comprovam a capacidade da empresa de produzir o produto e os requisitos terapêuticos, que comprovam a segurança e a eficácia do uso do produto. Segue adiante uma análise da legislação para o registro de medicamentos fitoterápicos, mostrando sua evolução e seus requisitos atuais.
Breve Histórico
As primeiras normas para regular de alguma forma a produção e comercialização de fitoterápicos foram os regimentos portugueses de 25/2/1521 e 12/12/1631. Estas normas abordavam a relação entre os agentes de saúde, o Estado e os usuários, e valiam tanto em Portugal quanto em suas colônias. Mesmo sem nunca terem sido implementadas na prática, estas duas normas continuaram valendo no Brasil, até a chegada da família real Portuguesa em 1808, quando foram promulgados o alvará de 23/ 11/1808 e a lei de 30/8/1828, que regularizavam a profissão de boticário, estabelecendo parâmetros de comportamento e práticas de produção (HENRIQUES, 1992; SIMÕES et al., 2003). Durante todo este período, até a publicação da primeira edição da Farmacopéia Brasileira, em 1929, não houve uma publicação oficial de referência nacional, ficando o Codex Medicamentarius Gallius como única obra de referência para a área. A primeira edição da Farmacopéia Brasileira foi um marco significativo no esforço de se regulamentar a manipulação de produtos de origem vegetal. Elaborada por Rodolfo Albino, ela incluía duzentas e oitenta espécies botânicas nativas e introduzidas. É uma obra importante, pois trata tanto de aspectos de controle da qualidade quanto de produção, refletindo as características da época - quando a maioria dos medicamentos era originária de plantas medicinais ou produtos biológicos (SIMÕES et al., 2003).
Em seguida à publicação da primeira edição da Farmacopéia Brasileira, foi promulgado o Decreto 19.606 de 19/1/1931 (BRASIL, 1931a), posteriormente regulamentado pelo Decreto 20.377 de 8/9/1931 (BRASIL, 1931b). Estes marcavam o início formal das atividades de Vigilância Sanitária no País, incluindo a estrutura do sistema, as responsabilidades pela fiscalização do exercício da Farmácia, além de regulamentarem várias questões da profissão farmacêutica como diplomas, instalação de farmácias, âmbito profissional, substâncias controladas, indústria farmacêutica e drogarias. Especificamente sobre plantas medicinais, o Decreto 19.606 determinava que o comércio de plantas medicinais de aplicações terapêuticas era privativo de farmácias e drogarias e as ervanarias só teriam a licença mantida até a mudança de propriedade, quanto teriam a licença cassada . Além disso, o decreto 20.377 determinava que era proibida a venda em ervanarias de produtos que se relacionassem com fetichismo ou curandeirismo e que o condicionamento de plantas ou partes vegetais deveria ser feito em recipientes adequados, fechados, livres de pó e contaminação. Por fim determinava que seriam apreendidas e inutilizadas as plantas sob classificação botânica falsa, bem como as desprovidas de ação terapêutica que fossem vendidas nestes estabelecimentos com nome vulgar de plantas ativas, além de prever a punição do infrator. Observa-se que até então as informações requeridas para a concessão da licença eram muito parcas. Apesar de abordar as questões principais como composição, indicação terapêutica, modo de preparo e controle da qualidade, estas informações eram solicitadas de forma vaga e para todas as especialidades farmacêuticas em geral. Especificamente em relação a plantas medicinais, observava-se uma preocupação positiva com seu uso científico, desvinculando-o de crenças religiosas. Até esse momento não se constata nenhuma legislação específica para medicamentos fitoterápicos, um fato compreensível para uma época na qual não existia uma distinção clara entre medicamentos de origem natural ou sintética.
Com o avanço do conhecimento científico e do desenvolvimento técnico, fica clara a necessidade de uma legislação que fosse específica para fitoterápicos. Como resultado, ocorre a promulgação da Portaria nº 22 de 30/10/1967 pelo Serviço Nacional de Fiscalização da Medicina e da Farmácia, que então estabelece normas para o emprego de preparações fitoterápicas. O primeiro destaque da norma é a apresentação de uma definição de produto fitoterápico, um conceito importante para a discriminação do fitoterápico e das demais especialidades farmacêuticas.
Outro avanço foi a inclusão, no ato de concessão da licença, da apresentação de um relatório em que constassem informações detalhadas sobre o produto. O relatório deveria conter informações sobre a produção e o controle da qualidade (requisitos tecnológicos) e de eficácia e segurança (requisitos terapêuticos). Entre os primeiros, destaca-se a caracterização botânica e farmacognóstica das drogas vegetais, indicando um controle de matéria-prima, além de informações sobre o controle da qualidade do produto, indicando suas características físico-químicas, ensaios de doseamento e atividade. No caso destas informações constarem da Farmacopéia Brasileira, fazia-se necessário apenas apresentar a monografia respectiva. Para os requisitos terapêuticos, destacase a apresentação de trabalhos científicos que corroborassem a indicação terapêutica. No caso de produto não constante da Farmacopéia, era necessária a apresentação dos resultados de ensaios farmacológicos e clínicos, que incluíam a toxidade imediata e tardia, ensaios pré-clínicos e clínicos e testes de teratogenicidade. A Portaria nº 22 ainda aponta que deveriam ser evitadas fórmulas com mais de uma droga vegetal, evitando as panacéias. A norma exigia estas informações não só de produtos novos mas também de produtos já licenciados, determinando um prazo para a apresentação das informações. Também ampliava sua abrangência para fitoterápicos empregados em homeopatia, medicamentos não contemplados em nenhuma legislação anterior. Portanto, a legislação abordava os pontos cruciais do setor e é, por si, um marco importante. Porém, a legislação era ainda limitada, não apresentando definições senão a de produto fitoterápico, e permanecendo muito vaga na apresentação de seus requisitos (MS, 1967; SIMÕES et al., 2003 ).
Paralelamente a este contexto, em 1959, foi publicada a segunda edição da Farmacopéia Brasileira, na qual, sob o argumento de ausência de ação terapêutica ou mesmo desuso das drogas, foram excluídas quinhentas monografias, representando cerca de duzentas espécies vegetais expressivas da nossa flora (SIMÕES et al., 2003). Nesta inflexão estabelece-se a tendência da Farmacopéia em tratar principalmente de fármacos sintéticos, refletindo o momento histórico no qual estes produtos passam predominar no mercado.
Na década de 70 são promulgadas as Leis nº 5.991 de 17/12/1973 e nº 6.360 de 23/09/1976. A Lei nº 5.991 dispôs sobre o controle sanitário do comércio de drogas, medicamentos e outros produtos, indicando que a dispensação de plantas medicinais é privativa de farmácias e ervanárias. Isto contrariava o Decreto 19.606 no que se refere à exclusividade da venda de plantas medicinais por farmácias e drogarias. Contudo, é interessante observar que todos os instrumentos regulatórios zelavam pelo acondicionamento adequado e a classificação botânica (BRASIL, 1973). A Lei nº 6.360 estabeleceu legalmente um sistema de vigilância sanitária de medicamentos, cosméticos, saneantes, insumos, correlatos e outros. Ela atualiza e consolida normas anteriores e cria, para medicamentos, a figura do registro, suprimindo o caráter definitivo da licença (BRASIL, 1976). O registro tem como vantagem o estabelecimento de um prazo de renovação, podendo ser cassado em caso de interesse público, sempre que houvesse fundamento para tal. A Lei nº 6.360 não aborda especificamente medicamentos fitoterápicos, o que levou ao surgimento de uma corrente que afirma que, para estes produtos, não se aplicaria esta legislação, apontando a identificação destes produtos como alimentos e gerando a polêmica observada até hoje com relação aos suplementos alimentares. A Lei nº 6.360 dá os primeiros passos para o controle sanitário da produção e da comercialização de medicamentos, mas (até por ser uma lei) não determina normas específicas, pelo contrário, indica que estas serão estabelecidas pelos agentes de vigilância sanitária responsáveis (BRASIL, 1976). Em 1992 foram publicadas duas normas que, apesar de bem específicas, tiveram grande importância: a Portaria SNVS nº 19, de 30/01/1992, e a Resolução nº 19 - SESA/PR de 10/03/1992. A primeira proibiu o uso da planta “confrei” em preparações para uso interno e a segunda proibiu o uso do “cambará”, tanto in natura como em formas farmacêuticas. Estas normas foram o resultado de avaliações técnicas sobre a toxidade de fitoterápicos, e colocou em questão a visão de que fitoterápicos não seriam tóxicos (SIMÕES et al., 2003).
Legislações recentes para o registro de medicamentos fitoterápicos
A partir de 1995 ocorre a publicação de uma seqüência de normas sobre o registro de fitoterápicos, alternando-se em intervalos de 5 anos em média. É interessante notar como foram sendo incorporados conceitos modernos sobre indústria farmacêutica, assim como a evolução dos requisitos tecnológicos e terapêuticos para o registro de um fitoterápico.
Portaria n° 6 de 31 de janeiro de 1995.
Já a partir de 1972, percebeu-se a necessidade de se atualizar a Portaria nº 22, principalmente pela lacuna deixada pela Lei nº 6.360. Em 1994, foi aberta uma proposta à consulta pública, de normatização para registro de medicamentos fitoterápicos. Mais tarde, após a avaliação das sugestões apresentadas, publicou-se a Portaria nº. 6 de 31/01/1995 (SIMÕES et al., 2003). A portaria n° 6 foi um grande avanço na normatização de medicamentos fitoterápicos. Ela introduz conceitos e determina requisitos tecnológicos e terapêuticos, numa clara evolução da Portaria n° 22.
Definições - A primeira parte da Portaria é a seção ‘definições’, inexistente na Portaria nº 22 (que inclui apenas a de medicamento fitoterápico), mas já de acordo com a lei nº 6.360 que tem como uma de suas principais virtudes o estabelecimento de conceitos. Esta seção é, portanto, importante pela introdução de conceitos claros sobre algumas particularidades da produção de fitoterápicos. Segue a definição de fitoterápico da Portaria n° 6:
Produto fitoterápico : é todo medicamento tecnicamente obtido e elaborado, empregando-se exclusivamente matérias-primas ativas vegetais com finalidade profilática, curativa ou para fins de diagnósticos, com beneficio para o usuário. É caracterizado pelo conhecimento da eficácia o dos riscos de seu uso, assim como pela reprodutibilidade e constância de sua qualidade: é o produto final acabado, embalado e rotulado.
Na sua preparação, podem ser utilizados adjuvantes farmacêuticos permitidos pela legislação vigente. Não podem estar incluídas substâncias ativas de outras origens, não sendo considerado produto fitoterápico quaisquer substâncias ativas, ainda que de origem vegetal, isoladas ou mesmo suas misturas.
Como se pode ver, a definição é elaborada e bastante completa, abordando todos os itens descritos na normativa, reafirmando, por um lado, a origem vegetal da matéria prima descrita na Portaria n° 22 e, por outro, conferindo exclusividade a esta origem, ao proibir claramente a adição de substâncias de outras origens. Quando estabelece que “...é caracterizado pelo conhecimento da eficácia e dos riscos de seu uso, assim como pela reprodutibilidade e constância de sua qualidade...” acaba por determinar de forma sucinta, os requisitos terapêuticos e tecnológicos que devem cumprir estes medicamentos. Obriga ao conhecimento científico sobre a farmacologia e a toxicologia (informações muitas vezes não comprovadas cientificamente) e, ainda, a garantir a qualidade do produto através de ferramentas de controle da produção. Chama a atenção o importante fato de atribuir aos fitoterápicos a mesma finalidade de todo medicamento, como descrita na Lei n° 6.360, o equiparando pois medicamentos sintéticos. Por outro lado determina que substâncias ativas de origem vegetal, isoladas ou mesmo em misturas, não são considerados fitoterápicos, numa clara alusão de que estes devem seguir o registro normal de medicamentos. Complementando a definição de fitoterápico, há as definições de matéria-prima vegetal, droga vegetal, e preparado fitoterápico intermediário.
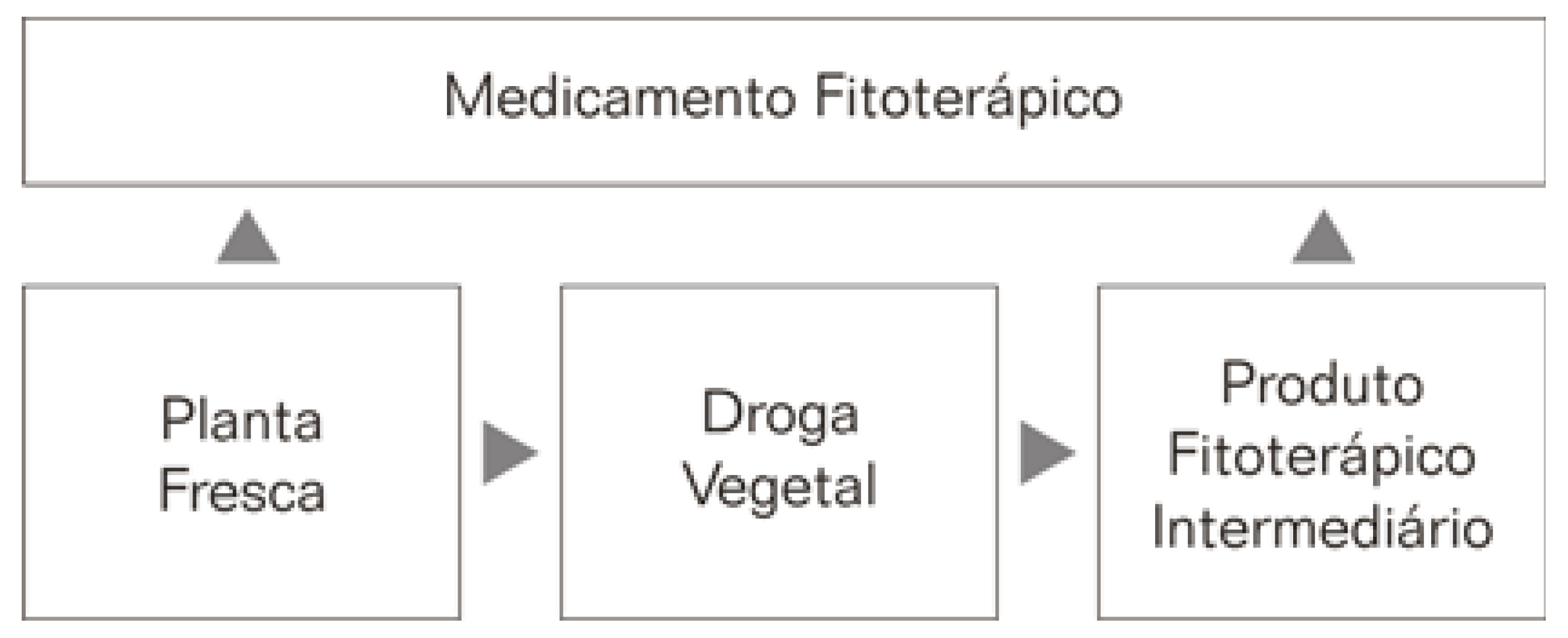
A droga vegetal é “...a planta ou partes, que após processo de coleta, secagem, estabilização e conservação justificam seu emprego na fabricação de medicamentos”. Já o produto fitoterápico intermediário é “...produto vegetal triturado, pulverizado, rasurado, extrato, tintura, óleo fixo, ou volátil, cera, suco e outros obtidos de plantas frescas e de drogas vegetais, através de operações de fracionamento extração, purificação ou concentração utilizado na preparação de produto fitoterápico”. Finalmente, a matéria prima vegetal é “...a planta fresca, droga vegetal ou preparado fitoterápico intermediário empregado na fabricação de produto fitoterápico”. Como se vê, as três definições descrevem os produtos intermediários do processo de produção do fitoterápico.
Como mostra a FIGURA 1, segundo a definição de matéria-prima vegetal, cada tipo de matéria-prima é uma etapa no processamento da planta para a produção do medicamento. Sendo que se pode produzir o medicamento fitoterápico a partir de qualquer um dos três tipos de matéria-prima.
A Portaria apresenta ainda duas definições importantes para a garantia e o controle da qualidade da produção de fitoterápicos: os conceitos de marcador e princípio ativo. Segundo a norma, princípio ativo é a “...substância ou grupo delas, quimicamente caracterizadas, cuja ação farmacológica e responsável, total ou parcialmente, pelos efeitos terapêuticos do produto fitoterápico” e marcadores “...são constituintes quimicamente definidos, presentes na matéria prima vegetal, preferencialmente os próprios ativos, destinados ao controle de qualidade da matéria-prima vegetal, dos preparados fitoterápicos intermediários e dos produtos fitoterápicos”. São duas definições intimamente ligadas e está claro que em ambos os casos são substâncias presentes no vegetal, sendo também cientificamente caracterizadas. A grande diferença é que o marcador é a substância escolhida para um determinado vegetal que pode ser determinada para atestar a qualidade do produto. Ao contrário dos medicamentos sintéticos, o marcador pode ser ou não o principio ativo (quando não se define cientificamente qual é o princípio ativo ou quando não há um método para se dosá-lo com a validade necessária). Assim, o marcador pode apenas atestar a qualidade da planta em si. Plantas com uma quantidade mínima de determinado marcador podem ser consideradas de boa qualidade e fornecedoras potenciais de matéria-prima com boa atividade farmacológica.
Registro de Fitoterápicos - Os requisitos inseridos na seção principal da norma explicitam que condições os produtos fitoterápicos devem cumprir para ter seus registros aceitos, condição para a permissão de produção e venda. Os requisitos podem ser divididos em requisitos tecnológicos, relacionados à produção e ao controle da qualidade; e requisitos terapêuticos, relacionados à segurança e eficácia do uso do medicamento. Os requisitos dos primeiros abordam o produto de cada etapa da produção do fitoterápico como definidas anteriormente: a planta fresca, o droga vegetal, produto fitoterápico intermediário e produto fitoterápico. Da planta fresca pede-se nomenclatura científica e popular junto com um laudo do profissional que atesta a identificação. Deve-se indicar a parte utilizada bem como se foi cultivada ou não o que, neste caso, obrigaria a apresentação de documentos que comprovem a aquisição da matéria-prima nos moldes da legislação ambiental. Por fim, como ferramentas de controle da qualidade constituem a caracterização farmacognóstica, os resultados de testes farmacopéicos para matérias-primas vegetais, a análise dos princípios ativos (quando possível) e uma análise fitoquímica qualitativa dos constituintes da espécie. Seguindo a ordem da produção, o passo seguinte é a droga vegetal. Os requisitos para ela são exatamente os mesmos da planta fresca, adicionando-se o método de estabilização - o que faz sentido, já que esta é a diferença entre estas duas etapas. Para o produto fitoterápico intermediário deve-se atender aos requisitos já descritos e adicionar a forma de preparo e suas características físico-químicas. O último item dos requisitos tecnológicos trata dos requisitos para o produto fitoterápico. Para este é necessário apresentar todos os dados de produção, como a fórmula do produto que será comercializado, a concentração da matéria-prima vegetal indicando a correspondência com os princípios ativos (quando possível), dados relacionados à produção; critérios para identificação de lote, processo de fabricação, assim como dados relacionados ao controle da qualidade como pontos de controle em processo; metodologias de controle físico-químico ou farmacológico (quando for o caso), além de ensaios de estabilidade com a determinação do prazo de validade. Trata-se, portanto, de estabelecer uma rastreabilidade desde a planta fresca até o produto final, visando desta forma, garantir a qualidade do produto acabado.
A divisão entre as diferentes etapas levanta a questão da eventual possibilidade de se registrar a matéria-prima. A norma não prevê essa possibilidade, mas também não o nega explicitamente. É importante também a exigência sobre a observação da legislação ambiental, mormente quando a planta não pode ser cultivada - nestes casos a planta é geralmente nativa.
Para os estudos de estabilidade não há a determinação de uma metodologia a ser adotada, deixando para o fabricante a escolha do caminho a ser seguido. No geral, observa-se que os requisitos para o produto fitoterápico são compatíveis com os da produção industrial farmacêutica. Os requisitos tecnológicos foram largamente ampliados e detalhados nesta norma, quando comparados com a Portaria nº. 22, refletindo uma nítida evolução científica e tecnológica.
Seguindo o processo de registro, a norma trata dos requisitos terapêuticos. Esta seção é muito simples e direta. Para se registrar um medicamento fitoterápico deve-se, primeiro, apresentar estudos científicos que comprovem a segurança do uso do produto acabado abordando toxicologia pré-clinica e clínica, com o estabelecimento da relação doseatividade, definir as indicações terapêuticas e dados sobre reações adversas, contra-indicações e restrições de uso. A norma determina ainda que todos os estudos devem ser realizados segundo as normas de pesquisa em saúde do Conselho Nacional de Saúde. Este ponto da norma é importante, pois, na prática, exige dos medicamentos fitoterápicos os mesmos requisitos dos medicamentos sintéticos. Em comparação com a Portaria nº 22, não há uma mudança nos requisitos em si, porém uma ampliação da sua aplicação, já que esta norma apenas era clara quanto aos estudos referentes aos produtos não oficinais. Para os produtos farmacopéicos era apenas necessário apresentar os trabalhos científicos que embasassem a indicação terapêutica sem, necessariamente, discriminar se se tratava de ensaio clínico. As novas exigências terapêuticas trouxeram um problema: boa parte dos medicamentos fitoterápicos no mercado não cumpria estas exigências. Para solucioná-lo, determinou-se, levando-se em conta o prazo de 5 anos de validade do registro determinado na Lei n° 6.360, que os produtos que já fossem registrados teriam o prazo igual para apresentar os resultados de segurança requeridos na norma, assim como 10 anos para apresentar a conformidade da eficácia segundo a norma. A rigor, isso implicou no problema de deixar no mercado produtos que não tinham os requisitos terapêuticos comprovados. Para amenizar esta situação, determinou-se a inclusão da seguinte inscrição na bula e nos rótulos dos produtos: “Produto em estudo para avaliação científica das indicações terapêuticas e da toxidade o uso deste produto esta baseado em indicações tradicionais” - o que permitia que o consumidor fizesse uma escolha consciente. Naturalmente que a norma ainda determina que caso fosse se constatado algum indício de toxidade neste período, o produto seria imediatamente retirado do mercado.
Isenção de Registro - Mesmo com o sistema de cumprimento de exigências, a norma ainda prevê a isenção de registro no caso de fitoterápicos que fossem descritos em monografias na Farmacopéia Brasileira ou em outro código oficial aceito. Ainda assim, era necessário apresentar todos os requisitos tecnológicos e terapêuticos que não constam na monografia, para completar a conformidade com a norma de registro.
Vale ressaltar que a isenção de registro é apenas uma forma de facilitar a aprovação de produção do produto. De qualquer forma deve-se cadastrar o produto junto ao órgão de vigilância sanitária.
Similares - Uma grande inovação desta norma foi quanto ao registro de similares. Não abordado na legislação anterior, o registro de similar tornavase agora permitido, contanto que se mantivesse a fórmula do fitoterápico de referência, com o mesmo princípio ativo ou matéria-prima, e que se apresentasse a metodologia para o controle da qualidade. Contudo, a grande inovação foi a inserção do requisito de testes clínicos que comprovassem a bioequivalência com o medicamento de referência, ou testes in vitro, desde que comprovada sua correlação com a biodisponibilidade. Obviamente, isso dificultou muito o registro de similares. Os fitoterápicos que não tivessem princípios ativos determinados, por exemplo, não poderiam fazer testes de bioequivalência e, mesmo quando houvesse, além das dificuldades normais de um ensaio de bioequivalência haveria ainda a influência da matriz do vegetal, que poderia ser diferente da referência, segundo a forma de processamento. É interessante notar que os medicamentos similares são obrigados hoje em dia, a apresentarem resultados de ensaios de bioequivalência, o que não ocorria na época. Vale ainda lembrar que esta Portaria data de 1995, bem antes da criação da Anvisa e da Lei dos genéricos.
Bulas e Rotulagem - Em relação à rotulagem e bulas, a Portaria detalha o que estas devem conter, como a designação “Produto Fitoterápico” e a nomenclatura oficial da planta; assim como não devem conter dizeres que induzam à automedicação ou levem a acreditar na segurança do produto somente por ser de origem vegetal. Devem também conter itens gerais de bulas como número de lote, nome da empresa, responsável técnico, e demais itens da legislação vigente.
Validação - A portaria insere pela primeira vez a exigência de validação de metodologias de controle da qualidade, apesar disso estar restrito a medica22 Revista Fitos mentos não farmacopéicos. É importante notar que, assim como para os estudos de estabilidade, não há menção à metodologia de validação, apenas a afirmação de que deve ser feita.
Associação de espécies - Ao contrário da Portaria nº 22, a Portaria nº 6 (MS, 1995) permite a associação entre espécies vegetais. Porém, estas associações devem estar embasadas em estudos científicos, garantindo a segurança e a estabilidade da associação e devem produzir efeitos adversos de igual ou menor intensidade que seus componentes separadamente.
Por fim, a Portaria determinava que à qualquer questão não prevista nesta norma seria aplicada a legislação de medicamentos, reforçando assim a indicação de regulação de fitoterápicos pela legislação desta classe de produtos (MS, 1995).
A Portaria n° 6 representou uma grande evolução no registro de fitoterápicos, ao melhor definir os conceitos relativos à indústria de fitoterápicos, organizando os requisitos tecnológicos segundo a produção, aí aportando lógica e ordem, ao melhor detalhar os requisitos tecnológicos e terapêuticos, atualizando-os pela evolução científica; além de definitivamente introduzir conceitos da moderna indústria farmacêutica, como os estudos de estabilidade, validação e bioequivalência, mesmo fora do contexto dos medicamentos genéricos.
Resolução RDC n° 17 de 24 de fevereiro de 2000
Com a criação da Anvisa, e devido à aproximação do fim do prazo de 5 anos para que todo medicamento fitoterápico apresentasse os resultados de segurança, iniciou-se um processo de revisão da Portaria nº 6, motivado principalmente pela dificuldade de adequação dos medicamentos às suas exigências quanto aos requisitos terapêuticos. O resultado foi a edição da Resolução RDC n° 17 em 24 de fevereiro de 2000, que trazia regulamento técnico sobre o registro de fitoterápicos (MS, 2000). Calcada na Portaria 06/95, a RDC 17 trazia algumas mudanças, principalmente no tocante aos requisitos terapêuticos, assim como o aprimoramento de algumas definições e requisitos tecnológicos. Em seguida analisam-se algumas diferentes seções desta resolução comparadas com a norma anterior.
Definições - Nesta seção existem algumas mudanças significativas de normas, que já antecipam algumas outras que aparecem adiante. A primeira mudança é a adição da definição de adjuvante como “...substância adicionada ao medicamento com finalidade de prevenir alterações, corrigir e/ou melhorar as características organolépticas, biofarmacêuticas e tecnológicas do medicamento”. Na portaria nº 6 não havia esta definição, embora o uso de adjuvantes estivesse previsto na definição de fitoterápico.
Seguindo com as matérias-primas, foi retirada a definição de “preparado fitoterápico intermediário”. Para substituí-la, foram adicionados à definição de matéria-prima exemplos que antes estavam descritos como naquela definição. Além disso, a RDC nº 17 passava a considerar produto vegetal triturado, rasgado e pulverizado como “droga”, em substituição ao que a Portaria nº 6 considerava como preparado “fitoterápico intermediário”. Sobre a definição de medicamento fitoterápico, a primeira modificação foi justamente a de passar a se chamar “medicamento”, e não mais “produto” (como na Portaria nº 6), reforçando o seu caráter terapêutico. A definição em si não apresenta muitas modificações, constando apenas da retirada da parte que tratava de adjuvante. Isso acarretava um pequeno problema já que, como estava a definição, determinava o uso apenas de matérias-primas vegetais, excluindo os adjuvantes de outras origens. Ainda sobre o conceito de medicamento, a norma introduz três novas definições, apontando quais são as principais modificações desta forma em relação à Portaria n° 6. São elas:
Medicamento fitoterápico novo - “...aquele cuja eficácia, segurança e qualidade, sejam comprovadas junto ao órgão federal competente, por ocasião do registro, podendo servir de referência para o registro de similares”.
Medicamento fitoterápico tradicional - “...aquele elaborado a partir de planta medicinal de uso alicerçado na tradição popular, sem evidencias, conhecidas ou informadas, de risco à saúde do usuário, cuja eficácia é validada através de levantamentos etnofarmacológicos e de utilização, documentações tecnocientíficas ou publicações indexadas”.
Medicamento fitoterápico similar - “...aquele que contém as mesmas matérias-primas vegetais, na mesma concentração de princípio ativo ou marcadores, utilizando a mesma via de administração, forma farmacêutica, posologia e indicação terapêutica de um medicamento fitoterápico considerado como referência”.
Assim, a norma divide o registro de medicamentos fitoterápicos em três classes, com diferentes requisitos terapêuticos para cada uma. Por fim, as definições de princípio ativo de marcador são mantidas praticamente iguais, retirando-se apenas a menção à caracterização química de marcador.
Registro - Como então apontado pelas novas definições, o registro de medicamentos fitoterápicos passa a ser feito em três tipos de medicamentos diferentes: medicamentos novos, medicamentos tradicionais e medicamentos similares. Os três requerem o cumprimento dos mesmos requisitos tecnológicos. Comparados com a Portaria nº 6, há algumas modificações. Para a planta fresca, a identificação é possível por análise farmacognóstica quando houver especificações para tanto; por outro lado, quando se tratar de planta nativa, deve-se apresentar certificado de origem e documentação comprovando a origem do material, conforme a legislação de uso sustentado e de preservação do patrimônio genético. Para a droga vegetal, adicionou-se aos requisitos a apresentação dos controles do processo de estabilização. Com a supressão da definição de preparado fitoterápico intermediário, passaram a vigorar os derivados de droga vegetal, conceitos estes sem definição até então. Neste caso, a diferença em relação à Portaria n° 6, no que diz respeito ao preparado fitoterápico intermediário, era a exigência de apresentação de um relatório do controle da qualidade. Para os medicamentos, as mudanças constituíram na reunião de vários requisitos de produção e controle agrupados em um único item e agora mais bem organizados na forma de um relatório. A isso se somava a inclusão de um item separado sobre testes de estabilidade, detalhando um pouco mais os requisitos atribuindo-lhe maior importância, porém ainda sem definir as condições de realização do estudo.
A grande modificação introduzida pela RDC 17 estava nos requisitos terapêuticos, quando passavam a diferenciar as já mencionadas três possibilidades existentes para registro de medicamentos fitoterápicos. O medicamento fitoterápico novo exigia o cumprimento dos requisitos terapêuticos exatamente como já estava descrito na portaria nº 6, contendo ensaios clínicos e pré-clínicos de segurança e eficácia. A segunda forma é o registro como medicamento fitoterápico tradicional. Neste caso, a comprovação dos requisitos poder-se-ia dar por três formas:
• Presença em uma lista apresentada em anexo à RDC, onde eram relacionadas 13 plantas de uso tradicional com nome científico, parte usada, forma de uso, indicação terapêutica, dose diária e via de administração. Estas especificações deviam ser respeitadas integralmente, sendo apenas possível formular outras formas farmacêuticas com a obrigatoriedade da apresentação do cálculo de equivalência das doses e testes de dissolução quando for o caso.
• Através de um sistema de pontuação, no qual foram atribuídos pontos pela presença da planta medicinal em questão em obras de referência internacional. Estas foram discriminadas em uma lista em anexo à mesma RDC, e compunham três grupos, valorados de acordo com a pontuação de 3, 2 ou 1; sendo os pontos consignados de acordo com o grupo em que se classifica a obra. Além disso, permitia-se o acúmulo de 0,5 pontos para cada publicação sobre a eficácia do uso em alguma obra técnicocientífica não incluída na lista de obras de referência. Para se considerar como cumpridos os requisitos terapêuticos, a planta deveria acumular 6 pontos. Da mesma forma, caso apresentasse estudos clínicos comprovando eficácia e segurança, a planta receberia a pontuação 6.
• Apresentação de um levantamento bibliográfico, comprovando ausência de risco tóxico para o usuário, assim como a ausência de grupos ou substâncias químicas tóxicas; indicação de uso episódico ou para curtos períodos de tempo, coerência com relação às indicações terapêuticas propostas, indicação para doenças consideradas leves e com finalidade profilática e, por fim, comprovação do uso seguro por mais de 10 anos.
Além do já disposto acima, também foi facilitada a possibilidade de medicamentos já registrados, com tempo de comercialização superior a 30 anos, de atingirem o status de “medicamento tradicional”, cabendo-lhes automaticamente 3 pontos.
Registro de medicamento similar - Outra forma permitida de registro foi através do conceito de similaridade e, para isso, o produto deveria atender à legislação de similares. No entanto a menção à equivalência farmacêutica constante na portaria nº 6 foi retirada desta norma ficando o medicamento sujeito à legislação de similares em vigor na época.
Isenção - A isenção de registro era permitida basicamente nos mesmos moldes da Portaria nº 6, porém com uma diferença fundamental: enquanto que por esta portaria a isenção era concedida para aqueles produtos que tivessem monografias da espécie vegetal ou do produto fitoterápico descritas na Farmacopéia Brasileira ou outro código oficial aceito, na RDC 17 apenas os que tivessem a monografia da formulação poderiam requerer isenção. Este pode ser considerado um avanço legal, já que a isenção trata do medicamento e não da planta. Vale lembrar que todas as informações sobre requisitos tecnológicos ou terapêuticos que não constem da monografia farmacopéica deveriam ser apresentados segundo esta norma.
Prazo de revalidação de registro dos medicamentos registrados antes de 31/01/ 1995 - Em relação ao prazo instituído na Portaria nº 6, para a apresentação de dados sobre segurança por medicamentos registrados antes de 31/01/1995, a norma concedeu mais um ano (o prazo venceu no ano de sua publicação), possibilitando aos produtores optar por mudar a classe de medicamento, por exemplo, de medicamento novo para medicamento tradicional, ou realizar os estudos de segurança. Para aqueles que se mantivessem como medicamentos novos, a norma mantinha o prazo já fixado para apresentação dos resultados de estudos de eficácia para 31/01/2005, então já fixado na Portaria nº 6.
Rotulagem e bulas - Desta seção, foram retirados todos os itens constantes da Portaria nº 6 que não fossem específicos para medicamentos fitoterápicos, afirmando que a legislação geral de rotulagem de medicamentos deveria ser seguida para demais aspectos. Além disso, reafirmava a obrigação de incluir os dizeres sobre os estudos de toxidade e indicações terapêuticas já estipuladas na mesma Portaria e a colocar a inscrição “Medicamento Fitoterápico Tradicional” em rótulos e bulas de medicamentos desse tipo (MS, 2000).
Observa-se assim, a inclusão de algumas pequenas mudanças e reorganizações nos requisitos tecnológicos em relação à Portaria nº 6 sem, no entanto, mudar sua essência. Em relação aos requisitos terapêuticos, a legislação abriu uma nova forma de registro, dando oportunidade aos medicamentos considerados de uso tradicional. O uso das chamadas listas positivas é um instrumento comum, muito utilizado na Europa, para facilitar o registro de produtos reconhecidos pelo uso tradicional; e o sistema de pontuação abre a possibilidade de se produzir medicamentos a partir de bibliografias reconhecidas internacionalmente, valorizando o conhecimento acumulado durante anos em órgãos como a Organização Mundial da Saúde ou a Comissão E. Por outro lado, estas duas formas de comprovação favorecem o registro de plantas estrangeiras, por estarem mais bem documentadas. A terceira possibilidade permite que medicamentos baseados em plantas de baixo risco e com tradição de uso seguro, possam ser registrados, numa indicação clara para a mudança de classe de medicamento destes produtos. O registro de similares teve uma queda de qualidade significativa com a retirada da menção de bioequivalência, provavelmente pelas dificuldades de se realizarem tais estudos nestes casos.
Resolução RDC 48 de 16 de março de 2004
A legislação atualmente em vigor para o registro de fitoterápicos é a RDC nº 48 (MS, 2004e), publicada em 16 de março de 2004, que possui algumas mudanças em relação à RDC nº 17, a partir da introdução de algumas exigências processuais, da adoção de alguns requisitos comuns aos medicamentos de uma maneira geral e ao aprimoramento de alguns itens da legislação anterior.
Abrangência - A adição deste item é a primeira mudança desta legislação, identificando quais tipos de produtos são passíveis de registro. Aqui, o fato mais relevante é deixar claro que plantas e drogas de origem vegetal não são passíveis de registro.
Definições - Neste item introduzem-se algumas modificações. Por exemplo, a adição das definições de derivado de droga vegetal e de matéria-prima, conforme descritas indiretamente na Portaria nº 6 mas não definida na RDC 17. Em relação aos medicamentos fitoterápicos, há uma alteração radical, refletindo uma das principais mudanças da legislação. As definições de medicamentos fitoterápicos novos, tradicionais e similares foram retiradas, sendo mantida apenas uma definição para medicamento fitoterápico, que sofreu algumas modificações em relação à definição geral da Portaria nº 6, a qual foram incluídas as três formas de comprovação de requisitos terapêuticos introduzidas na RDC nº 17. Quando trata da exclusividade de matérias-primas vegetais, a normativa menciona matérias-primas ativas, contornando a pequena confusão da definição da Portaria nº 6. A menção sobre a finalidade, comum a todos os medicamentos foi suprimida, ressaltandose separadamente a definição de medicamento. Foi introduzida uma definição para “fórmula fitoterápica”, de acordo com o já descrito na legislação, e uma definição de “fórmula mestra”, não constante nas legislações anteriores: “...documento ou grupo de documentos que especificam as matérias-primas e os materiais de embalagem com as suas quantidades, justamente a descrição dos procedimentos e precauções necessárias para a produção de determinada quantidade de produto terminado. Além disso, fornece instruções sobre o processamento, inclusive sobre os controles de processo”. A fórmula mestra é um documento comum na indústria farmacêutica e a sua introdução reafirma o caráter industrial da produção de fitoterápicos.
À definição de marcador foi adicionada uma frase indicando a preferência por marcadores que tenham correlação com a atividade farmacológica, quando não forem os próprios princípios ativos; e a retirada da expressão “grupos de substâncias”, conforme descrito na portaria nº 6, colocando em seu lugar a expressão “classes de compostos químicos” - uma expressão menos abrangente e mais apropriada tecnicamente. Esta mesma alteração foi realizada na definição de princípio ativo.
Medidas antecedentes ao registro - Este item não existia na Portaria nº 6 e representa mais uma adaptação à legislação de medicamentos. Nele se descrevem a obrigação de notificar a produção de lotes pilotos, com base no guia apropriado da Anvisa (MS, 2006).
Registro - A alteração fundamental desta norma está justamente neste item, não havendo mais a divisão entre diferentes tipos de medicamentos, mas sim apenas uma divisão entre requisitos tecnológicos e terapêuticos. No tocante aos requisitos tecnológicos, há várias mudanças. A primeira que chama a atenção é a retirada do item “planta fresca”, permanecendo apenas os itens sobre “droga vegetal” “derivado de droga vegetal” e “medicamento fitoterápico”. Os requisitos para medicamentos são: apresentação de bula, indicação de prazo de validade conforme estudos de estabilidade acelerada segundo guia próprio da Anvisa (MS, 2005), relatório completo de produção, e relatório de controle da qualidade com descrição e validação das metodologias utilizadas segundo o guia da Anvisa (MS, 2003). Do item bula foi suprimida a determinação de não conter dizeres que levem à automedicação ou mesmo à sugestão de maior segurança, por ser de origem natural. O relatório de controle mencionado cumpre os mesmos moldes do relatório da Portaria nº 6, ao que foram adicionados os tamanhos mínimos e máximos dos lotes.
Para a droga vegetal, as mudanças são a requisição de metodologias validadas segundo guia próprio da Anvisa para drogas não farmacopéicas, e a retirada de testes de autenticidade e pureza e a exigência de comprovação da origem quando a planta fosse nativa. Para os derivados de drogas vegetais há poucas mudanças. A primeira é o requerimento de informações sobre solventes e/ou veículos utilizados na extração do derivado, junto com análise do fornecedor, a validação de métodos de análise pelo guia próprio da Anvisa, e a retirada, assim como na droga vegetal, da certificação de origem. A norma exige ainda especificações de material de embalagem e o certificado de Boas Práticas de Fabricação para a linha de produção. Devem-se enviar informações sobre o controle da encefalopatia espongiforme, quando for cabível. As associações de plantas, um assunto não mencionado na RDC n° 17, passa a ser permitido pela norma, contanto que se justifiquem com estudos e se apresentem evidências de uso tradicional. A RDC n° 17 determinava que os produtos importados deveriam seguir os mesmos requisitos dos medicamentos nacionais. Na norma atual foi adicionado um item que aprofunda um pouco a questão e fornece algumas orientações específicas para medicamentos importados, ainda que no geral, os requisitos continuem os mesmos que para os nacionais. Os requisitos terapêuticos - a principal mudança desta norma, na verdade, embutem a diferença entre cada tipo de medicamento fitoterápico descrito na RDC nº 17. Na atual norma, são apresentados como três formas diferentes de se comprovar a segurança e eficácia do uso do medicamento, com algumas modificações: tanto a lista de bibliografias como a lista de plantas para o registro simplificado, foram retiradas da norma principal e colocadas em normas separadas (MS, 2004a; MS, 2004b); para os ensaios de toxicologia pré-clínica deve-se usar como parâmetro o guia próprio da Anvisa e para os medicamentos de baixo risco, o período de comprovação de uso seguro aumentou de 10 para 20 anos.
Medidas pós-registro - A norma também prevê medidas pós-registro, como as alterações pósregistro, segundo guia próprio da Anvisa (MS, 2004c) e a entrega dos resultados finais de estabilidade de longa duração, assim como formar e comprovar a implantação de um sistema de farmacovigilância e os procedimentos legais para a renovação do registro (MS, 2004d).
A adoção dos guias padronizados da Anvisa certamente elevou a qualidade exigida nos requerimentos de registro para o mesmo nível dos medicamentos sintéticos, além de permitir diversas comparações úteis devido à uniformização dos requisitos. É importante notar que foram suprimidos: o registro por similaridade, e a isenção de registro. O fim da similaridade é um passo importante, pois não há como garantir a similaridade de medicamentos quando condicionados a diversos fatores como variabilidade de matéria-prima e diferenças de processos que eventualmente afetem a biodisponibilidade do(s) fármaco(s). A isenção de registro esteve sempre, desde as primeiras legislações, vinculada à existência de monografias do produto na Farmacopéia Brasileira. Duas questões são relevantes: primeiro, a isenção não se vinculava em um prazo para renovação, não prevendo, portanto, uma revalidação. Adicionalmente, a Farmacopéia cada vez mais se coloca como uma obra referência no controle da qualidade e, desta forma, a inclusão da monografia de um produto não pode mais servir de parâmetro para a isenção de registro, outrossim para complementar as informações sobre o controle da qualidade. Outro resultado importante, derivado do fim da classificação de medicamento fitoterápico tradicional, é a não obrigatoriedade de se escrever ‘medicamento’ no frasco.
Legislação Internacional
A legislação para o registro de fitoterápicos está em foco no restante do mundo. Com o aumento do uso de produtos à base de plantas pela população, a FDA americano (Food and Drug Administration) iniciou uma discussão sobre uma nova abordagem para o registro destes medicamentos (FDA, 2004). Paralelamente, a Europa discute a integração das legislações dos diferentes países que formam o bloco da Comunidade Européia. Assim como no Brasil, estas discussões têm por objetivo assegurar a qualidade (eficácia e segurança) dos produtos vendidos no mercado.
Nos EUA, as plantas medicinais são consideradas suplementos nutricionais, sujeitos ao Dietary Supplement Health and Education Act (DSHEA), de 1994. Esta legislação veio em resposta à crença de que plantas medicinais são seguras e de que a população desejava um maior acesso a estes produtos, que não estariam disponíveis no mercado se estivessem sujeitos às mesmas restrições dos medicamentos. O registro de medicamentos é um processo oneroso e demorado e um dos objetivos da legislação é permitir a produção e venda de fitoterápicos que podem ter efeito farmacológico sem ter, contudo, a extensa evidência necessária para registro como medicamento. É interessante notar que como não são obrigados a apresentar evidências científicas de eficácia, estes produtos não podem reivindicar indicações terapêuticas, apenas indicações nutricionais. Porém uma pesquisa recente nos sítios de diversos produtores na internet revelou que 55% deles reivindicavam ações terapêuticas (BENT; KO, 2004). A segurança é outro aspecto importante. O DSHEA determina que os fabricantes de suplementos nutricionais são responsáveis pela segurança de seus produtos, porém não são obrigados a apresentar nenhuma evidência de segurança ao FDA. Este, ao contrário, deve demonstrar que um produto não é seguro antes de poder retirá-lo do mercado. Esta é uma tarefa difícil; tomando-se em conta que os fabricantes não são obrigados a notificar os efeitos adversos dos seus produtos. Do ponto de vista da Vigilância Sanitária, esta legislação deixa o mercado sem um controle para garantir a saúde da população. A conseqüência direta e o risco deste descontrole podem ser evidenciados por um estudo realizado com 25 produtos à base de ginseng, que demonstrou uma variação de até 200 vezes em seu conteúdo em princípios ativos (BAST et al., 2002; BENT; KO, 2004). Em resposta a esta situação, o FDA publicou em junho de 2004 o Guidance for Industry Botanical Drug Product. Este documento fornece indicações sobre como registrar produto à base de matérias-primas vegetais, algas ou fungos microscópicos como medicamento, suplemento nutricional ou alimento, dependendo do uso específico.
Para medicamento, o documento descreve duas formas de registro, com ou sem prescrição médica, seguindo os mesmos procedimentos dos medicamentos sintéticos. Chama a atenção que o documento reconhece que muitos produtos derivados de matériasprimas vegetais são vendidos como suplementos nutricionais e, por isso mesmo, sem ensaios clínicos e que é importante avaliar a eficácia de tais produtos. Então permite que para estes casos, no quais também não se tenha reconhecido a ocorrência de qualquer problema com a segurança; as informações sobre farmacologia e toxicologia pré-clínica, necessárias para se iniciar um estudo de novo droga (Investigative New Drug - IND) será reduzida, em comparação com processos comuns de novas drogas. Afirma ainda que na maioria dos casos não serão necessárias informações adicionais sobre toxicologia ou requisitos tecnológicos, para se iniciar o processo de IND. Essa é uma clara iniciativa do FDA de tentar conduzir os suplementos nutricionais a sofrerem estudos mais aprofundados sobre eficácia do uso (FDA, 2004).
Na Europa, o uso de plantas medicinais com fins terapêuticos está profundamente inserido nos costumes da população. Com a consolidação da União Européia, os países membros passaram a seguir também a sua legislação, que estabelece a entrada no mercado europeu mediante a apresentação de documentação científica que comprove a qualidade, eficácia e segurança dos produtos farmacêuticos. A legislação estabelece também que todos os Estados membros devem checar se os produtos presentes atualmente no mercado cumprem os requisitos estabelecidos na Legislação Européia. No entanto, cada país tem a sua própria legislação sobre o registro de fitoterápicos, que levam em conta suas tradições e pontos de vista.
Para o registro de qualquer produto farmacêutico no mercado europeu, existem dois procedimentos possíveis: o centralizado, através da Agencia Européia de Avaliação de Medicamentos (EMEA), e o procedimento descentralizado, onde o registro é realizado individualmente em cada país, baseado no reconhecimento mútuo de registro entre países. Porém, caso algum país do bloco levante restrições sobre algum produto já registrado em outro país membro, as questões são resolvidas pela EMEA. Caso a Agência dê um parecer negativo, todos os registros já realizados nos diferentes países do bloco são perdidos. Produtos à base de plantas medicinais estão particularmente sujeitos a esta condicionante, pois as legislações e tradições dos diferentes países encaram os mesmos produtos de forma distinta, incluindo-se nisso as diferentes indicações terapêuticas. Estes tipos de obstáculos são preocupantes, pois podem impedir a harmonização da legislação para a área, causando problemas para a indústria e as autoridades (BAST et al., 2002; BENZI; CECI, 1997).
Desta forma, no tocante à legislação específica para fitoterápicos, cada país tem a sua abordagem, todavia é possível identificar pontos em comum. Na grande maioria dos países há mecanismos específicos para se contemplar medicamentos baseados no uso tradicional. Vários países possuem as chamadas listas positivas, nas quais se encontram aquelas plantas já com eficácia e segurança reconhecidas para determinadas indicações de uso tradicional. A lista geralmente especifica a forma farmacêutica a ser utilizada. Alguns países permitem que o registro de produtos à base de plantas medicinais seja obtido como alimento, como é o caso, por exemplo, de chás. Estes produtos, porém, não podem fazer nenhuma reivindicação terapêutica, pois teriam que ser então obrigatoriamente registrados como medicamento. Por outro lado, todos os países são exigentes quanto a um bom nível de qualidade para os processos de manufatura e industrialização, pautados pelas Boas Práticas de Fabricação e, em alguns casos, boas práticas agrícolas (MS, 2004a, BENZI; CECI, 1997).
Um caso específico que vale a pena ressaltar é o da Alemanha. Em 1984, aquele país criou um grupo especial de especialistas, constituindo a Comissão E, cuja função principal foi estabelecer os critérios de segurança e eficácia para plantas medicinais e suas preparações derivadas. Um fabricante apenas precisa apresentar as informações tecnológicas, confiando nas informações sobre eficácia e segurança constantes na monografia positiva da Comissão. Em 1994, uma nova lei estabeleceu que o ônus de provar eficácia e segurança dos produtos passa a ser do fabricante, apesar de ainda poder se referenciar em monografias da Comissão. No entanto, é muito difícil encontrar monografias para a grande maioria das plantas medicinais, além de que estas dificilmente abordam todas as formas farmacêuticas e usos possíveis, Há que se considerar o avanço da ciência e que novas indicações podem ser determinadas (BENZI; CECI, 1997).
Assim a legislação brasileira sobre registro de medicamentos fitoterápicos vem evoluindo paulatinamente, desde quando tratava apenas de plantas medicinais até os dias de hoje quando passa a tratar apenas de medicamentos. Nos últimos 12 anos houve uma evolução rápida com a publicação de três normas, reflexo deste período em que ocorreu a criação da Anvisa e a modernização por parte desta agência, não apenas da legislação de fitoterápicos, mas de todos os produtos sujeitos a vigilância sanitária. Os principais aprimoramentos realizados foram: o reconhecimento do fitoterápico como medicamento, com a aproximação dos seus requisitos aos dos medicamentos sintéticos, como a adoção dos guias de validação, estabilidade e lotes piloto, determinando não só a exigência destes estudos mas também a forma como eles devem ser feitos e a exigência de certificado de Boas Práticas de Fabricação e Controle para a linha de produção. Além disso, há a inclusão de métodos alternativos para a comprovação de requisitos terapêuticos, como a lista simplificada e o sistema de pontuação (instrumentos já utilizados em alguns países). Isso permite o registro de medicamentos à base de plantas tradicionalmente conhecidas e com segurança e eficácia comprovadas, de forma facilitada, apesar de na prática valer apenas para o registro de fototerápicos à base de plantas exóticas (‘medicamentos estrangeiros’). A Resolução RDC nº 48 é a norma atual em vigor e consolida os aperfeiçoamentos realizados que vão de encontro à legislação internacional, porém sempre admitindo futuros aprimoramentos.
Referências
. BAST, A.; CHANDLER, R. F.; CHOY, P.C.; DELMULLE, L.M.; GRUENWALD, J.; BART, S.; HALKES, A.; KELLER, K.; KOEMAN, J. H.; PETERS, P.; PRZYREMBEL, H.; et al. Botanical health products, positioning and requirements for effective safe use. Environmental Toxicology and Pharmacology, v.12, n.4, p.195-211, 2002.
. BENT, S.; KO, R. Commonly Used Herbal Medicines in the United States: A Review. The American Journal of Medicine, v.116, n.7, p.478-485, 2004.
. BENZI, G.; CECI, A. Herbal Medicines in European Regulation. Pharmacological Research, v.35, n.5, 355-362, 1997.
. BRASIL. Decreto n° 19.606 de 19 de Janeiro de 1931a. Dispõe sobre a profissão farmacêutica e seu exercício no Brasil. Brasil, 1931; apudAlves Filho, G.A.P.Vigilância Sanitária de Medicamentos no Brasil: uma análise da Legislação Sanitária Federal relativa à Responsabilidade Técnica de estabelecimentos de dispensação de Medicamentos. Monografia ENSP-FIOCRUZ, Rio de Janeiro, 2003. Disponível em: <http://www.direb.fiocruz.br/>
. BRASIL. Decreto n° 20.377 de 8 de setembro de 1931b. Atribuições da Profissão de Farmacêutico. Brasil, 1931. Disponível em (CRF): <http://www.crfsp.org.br/juridica/legislacao/legislacao>.
. BRASIL. Lei n° 5991 de 17 de dezembro 1973. Dispõe sobre o controle sanitário do comércio de drogas, medicamentos, insumos farmacêuticos e correlatos, e dá outras providências, Brasil, 1973. Disponível em <http://www.anvisa.gov.br/legis/leis/>.
. BRASIL. Lei n° 6360 de 23 de setembro 1976: Dispõe sobre a vigilância sanitária a que ficam sujeitos os medicamentos, as drogas, os insumos farmacêuticos e correlatos, cosméticos, saneantes e outros produtos, e dá outras providências, Brasil, 1976. Disponível em: <http://e-legis.bvs.br/leisref/public/>
. FDA: FOOD AND DRUG ADMINISTRATION, Guidance For Industry Botanical Drugs, Center for Drug Evaluation and Research, U. S. Department of Health and Human Services, July 2004, USA, 2004.
. HENRIQUES, C.M.P. A Vigilância Sanitária dos portos: A experiência de prevenção à entrada da cólera no porto de Santos. Tese de Mestrado, Faculdade de Medicina da Universidade de São Paulo, São Paulo, 1992.
. MS: MINISTÉRIO DA SAÚDE. Portaria n° 22 de 30 de outubro de 1967. Estabelece normas para o emprego de preparações fitoterápicas, Brasil, 1967. Disponível em: <http://e-legis.anvisa.gov.br/leisref/public/>.
. MS: MINISTÉRIO DA SAÚDE. Sistema de Vigilância Sanitária, Portaria nº 6 SVS/MS de 31 de Janeiro de 1995. Institui e normatiza o registro de produtos fitoterápicos junto ao Sistema de Vigilância Sanitária, Brasil, 1995. Disponível em: <http://e-legis.bvs.br/leisref/public/>.
. MS: MINISTÉRIO DA SAÚDE. Agência Nacional de Vigilância Sanitária, RDC nº 17 de 24 de Fevereiro de 2000. Dispõe sobre o registro de medicamentos fitoterápicos. Brasil, 2000. Disponível em: <http://e-legis.bvs.br/leisref/public/>.
. MS: MINISTÉRIO DA SAÚDE. Agência Nacional de Vigilância Sanitária, RE n° 899 de 29 de maio de 2003. Determina a publicação do “Guia para validação de métodos analíticos e bioanalíticos”; Brasil, 2003. Disponível em: <http://e-legis.anvisa.gov.br/leisref/public/>.
. MS: MINISTÉRIO DA SAÚDE. Agência Nacional de Vigilância Sanitária, RE nº 89 de 16 de Março de 2004. Determina a publicação da LISTA DE REGISTRO SIMPLIFICADO DE FITOTERÁPICOS, Brasil, 2004a. Disponível em <http://www.ibpm.org.br/legislacao>.
. MS: MINISTÉRIO DA SAÚDE. Agência Nacional de Vigilância Sanitária, RE n° 90 de 16 de março de 2004. Determina a publicação da GUIA PARA A REALIZAÇÃO DE ESTUDOS DE TOXICIDADE PRÉCLÍNICA DE FITOTERÁPICOS, Brasil, 2004b. Disponível em: <http://e-legis.anvisa.gov.br/leisref/public/>.
. MS: MINISTÉRIO DA SAÚDE. Agência Nacional de Vigilância Sanitária, RE n° 91 de 16 de Março de 2004. Determina a publicação do GUIA PARA REALIZAÇÃO DE ALTERAÇÕES, INCLUSÕES, NOTIFICAÇÕES E CANCELAMENTOS PÓS REGISTRO DE FITOTERÁPICOS, Brasil, 2004c. Disponível em: <http://e-legis.anvisa.gov.br/leisref/public/>.
. MS: MINISTÉRIO DA SAÚDE. Agência Nacional de Vigilância Sanitária, RDC n° 48 de 16 de março de 2004. Dispõe sobre o registro de medicamentos fitoterápicos, Brasil, 2004d. Disponível em: <http://e-legis.anvisa.gov.br/leisref/public/>.
. MS: MINISTÉRIO DA SAÚDE. Agência Nacional de Vigilância Sanitária, RE n° 1 de 29 de Julho de 2005. Autoriza ad referendum, a publicação do Guia para a Realização de Estudos de Estabilidade Brasil, 2005. Disponível em: <http://e-legis.anvisa.gov.br/leisref/public/>.
. MS: MINISTÉRIO DA SAÚDE. Agência Nacional de Vigilância Sanitária, RE n° 2999 de 12 de setembro de 2006. Determina a publicação do Guia para Notificação de Lotes-Piloto de Medicamentos, Brasil, 2006. Disponível em: <http://e-legis.anvisa.gov.br/leisref/public/>.
. SIMÕES, C.M.O.; FALKENBERG, M.B.; SANTOS, R.I. Introdução à análise fitoquímica. In: SIMÕES, C.M.O.; SCHENKEL, E.P.; GOSMANN, G.; MELLO, J.C.P.; MENTZ, L.A.; PETROVICK, P.R. (org.) Farmacognosia: da planta ao medicamento. 5.ed. Porto Alegre: Editora Universidade, UFRGS, 2003.