Estado da Arte
Produtos Naturais em Fase Avançada de Testes Clínicos no Tratamento contra o Câncer
Natural Products in Advance Clinical Trials Applied to Cancer
Resumo
Atualmente, os produtos naturais representam uma inestimável fonte de substâncias químicas tendo um importante papel no tratamento do câncer. Devido à importância da natureza como fonte de novos candidatos a fármacos no tratamento contra o câncer, o objetivo dessa revisão é enfocar os produtos naturais, que estão em fase avançada de testes clínicos e dentre em breve poderão ser introduzidos no mercado no combate ao câncer.
- Unitermos:
- Antineoplásica.
- Fármacos Anticâncer.
- Desenvolvimento de Fármacos..
Abstract
Nowadays, natural products represent an outstanding source of compounds that play an important role in cancer treatment. Due to the importance of nature as a source of new cancer drugs candidates, the aim of this review is to highlight natural products, which are under clinical trials against this disease.
- Key Words:
- Anti-neoplasic Activity.
- Anti-cancer Drugs.
- Drug Development..
Introdução
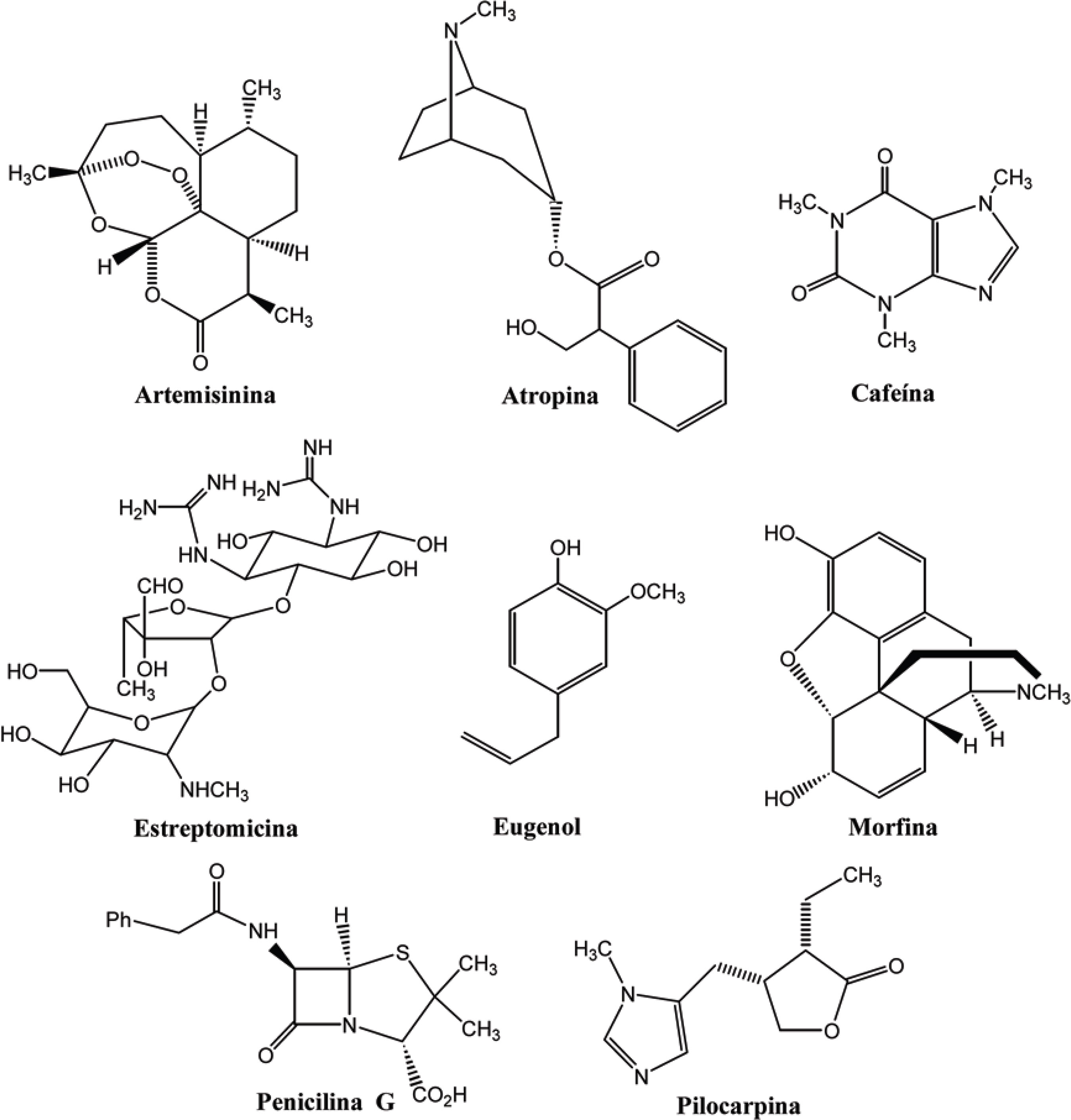
A utilização de plantas no tratamento de doenças no homem data dos tempos remotos de sua história, com registro na antiga china a cerca de 2700 A.C. e em papiros egípcios por volta de 1500 A.C.. No entanto, apesar da grande importância da humanidade pela natureza no decorrer da sua história, o desenvolvimento e aplicação dos produtos naturais apresentou pouco progresso até a década de 1940. Isto se deve às inúmeras limitações existentes para a caracterização dos princípios ativos presentes nas espécies, já que, até então, a principal ferramenta utilizada na elucidação dessas substâncias era a realização dos métodos químicos de análise, que consistiam no uso de reações para degradar, derivatizar e identificar grupos funcionais, na tentativa de se determinar a estrutura da substância desejada. Como conseqüência, eram necessárias grandes quantidades da substância a ser analisada. Isso, para a área de produtos naturais é uma séria limitação, já que, na grande maioria dos casos, as substâncias são isoladas em quantidades na ordem de miligramas. Apesar da importância dos métodos químicos de análise para o desenvolvimento da química, a área de produtos naturais somente pôde se desenvolver a partir do advento dos métodos físicos de análise e de purificação, como por exemplo, a espectroscopia na região do visível, ultravioleta e infravermelho, desenvolvidas no início da década de 1940, ressonância magnética nuclear na década de 1960, espectrometria de massas e cro- A importância dos produtos naturais, de origem vegetal ou animal, no combate a diferentes tipos de doenças adquiridas pela espécie humana, pode ser vista pelo grande número de fármacos que estão disponíveis no mercado. Como exemplo, podem-se citar: a artemisinina, antimalárico obtido da planta Artemisia annua; a atropina, anticolinérgico extraído de Atropa belladona; a cafeína, estimulante extraído de diferentes plantas, como Coffea arabica (café), Ilex paraguariensis (erva-mate) e Paullinia cupana (guaraná); a estreptomicina, o primeiro antibiótico utilizado no tratamento da tuberculose, isolado do fungo Streptomyces griseus; o eugenol, anestésico local, recomendado em casos de dor de dente, extraído do cravo-da-índia (Syzygium aromaticum); a morfina, narcótico utilizado como analgésico isolado da papoula (Papaver somniferum); a penicilina G, antibiótico isolado do fungo Penicillium notatum; e a pilocarpina, extraída das folhas da planta Pilocarpus jaborandi, sendo utilizada como colírio no tratamento do glaucoma (Figura 1) (ROCHA et al., 2001).
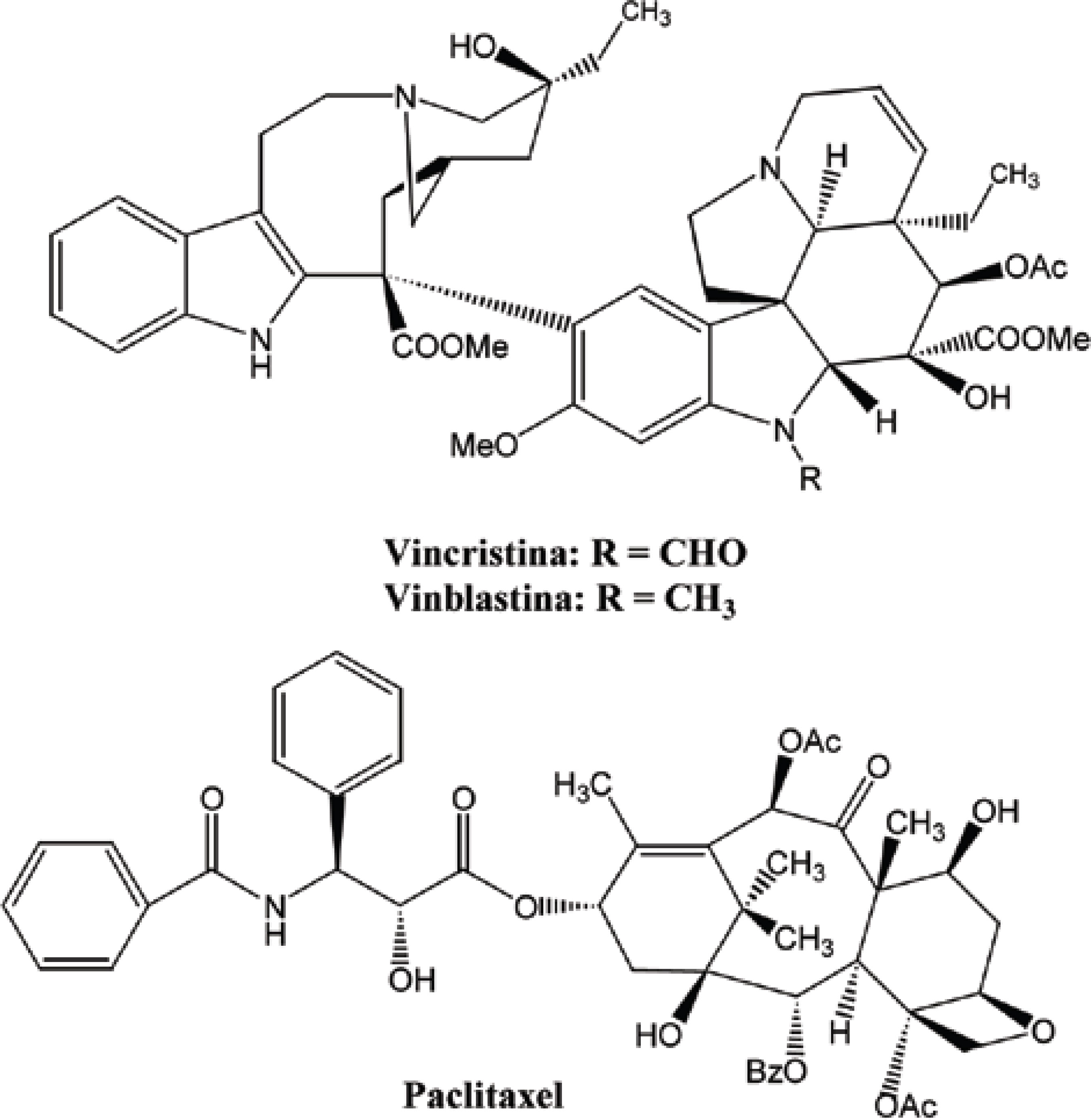
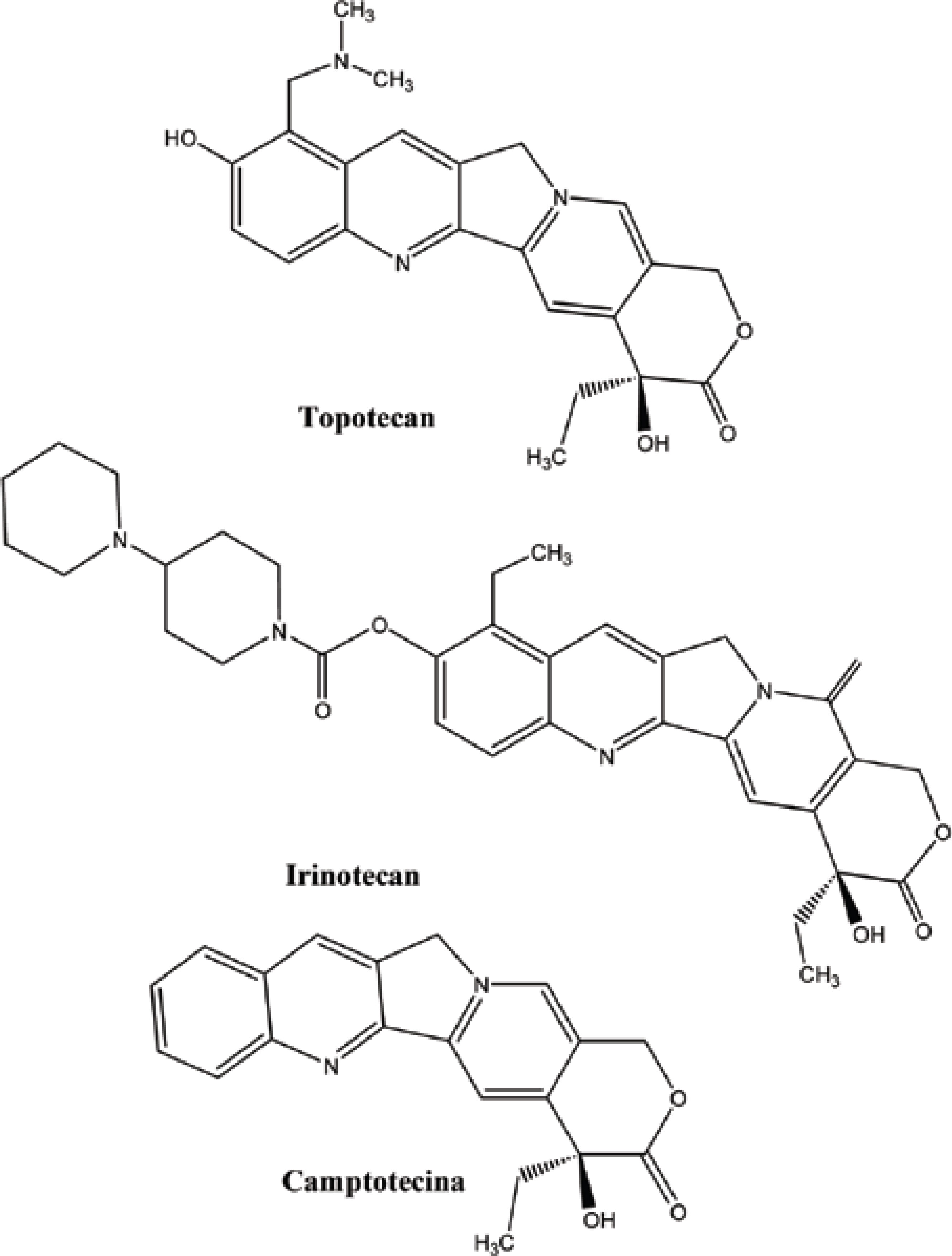
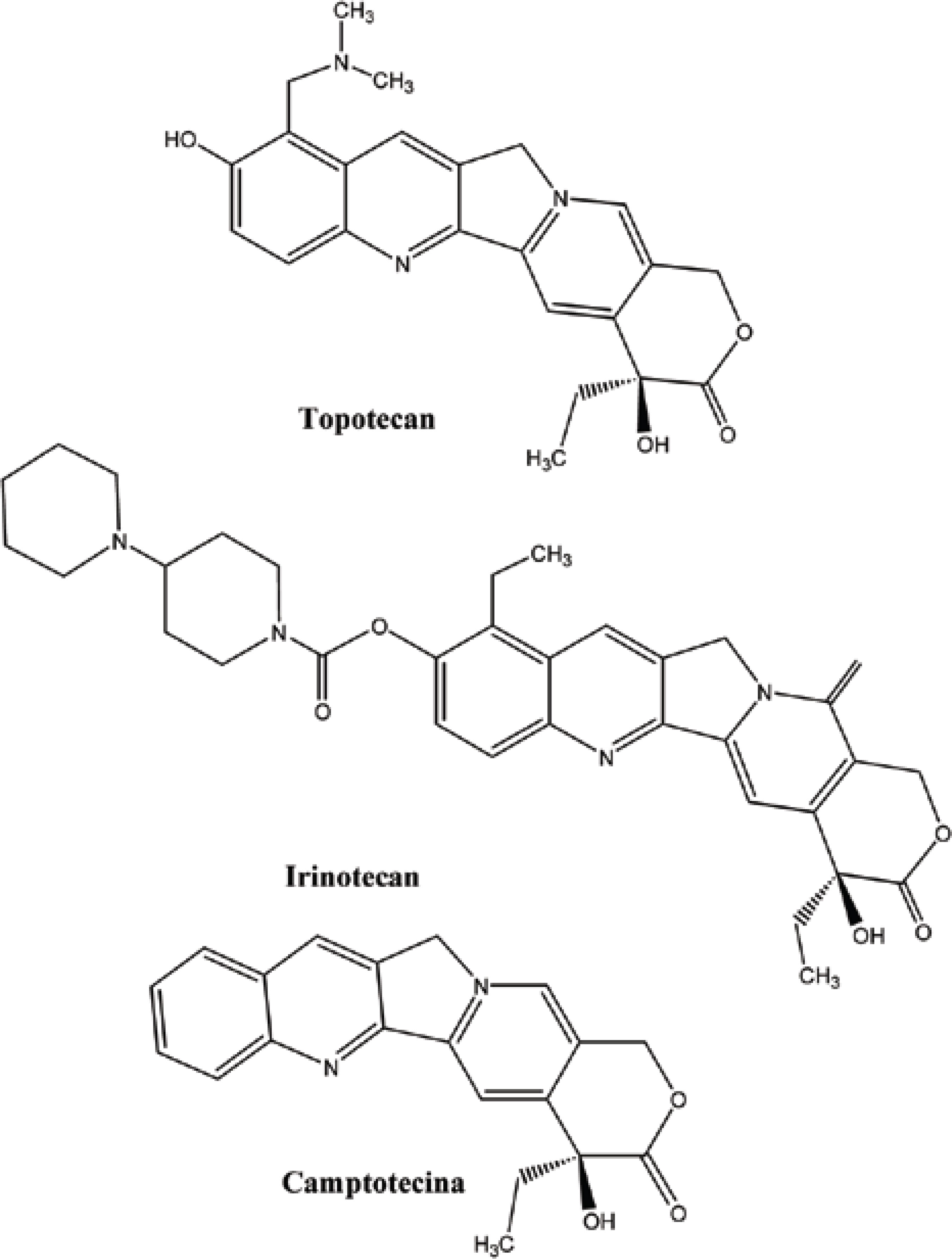
O câncer se apresenta hoje como uma das grandes preocupações da era moderna. Neste contexto, estudos de novos compostos com atividade antineoplásica, que têm como fonte produtos naturais, mostram-se cada vez mais uma importante ferramenta no combate a essa enfermidade. Fato esse que pode ser exemplificado a partir do número de tratamentos disponíveis, tendo como origem plantas medicinais. No período compreendido entre 1940 e 2002, 40% dos fármacos utilizados no tratamento do câncer eram derivados de produtos naturais (NEWMAN et al., 2003). Dentre esses se encontram a vinblastina e a vincristina, alcalóides extraídos da Catharanthus roseus, que participam do tratamento anticâncer a mais de 40 anos, e o paclitaxel, isolado do Taxus brevifolia, introduzido no mercado por volta dos anos 90 (Figura 2) (JULSING et al., 2006; SOUZA, 2004). Neste contexto, pode-se destacar também o Irinotecam e Topotecam, derivados semi-sintéticos da Camptotecina, um alcalóide quinolínico extraído da Camptotheca acuminata (Figura 3) (POMMIER, 2006) por Wall e colaboradores em meados da década de 60, que teve seus primeiros registros de atividade antineoplásica no início da década de 70.
Atualmente, cerca de 50% dos medicamentos encontrados no mercado para o tratamento de neoplasias ou têm princípios ativos de origem natural, ou são derivados semi-sintéticos. Isso torna evidente a importância da busca de novos fármacos com base nos estudos das substâncias que a natureza proporciona (MANN, 2002). Neste contexto, o objetivo desta revisão é evidenciar alguns produtos naturais que estão em fase avançada de testes clínicos, e em breve poderão ser utilizados na luta contra o câncer.
Estatísticas
Nas últimas décadas, tem se observado o desenvolvimento de novas enfermidades associadas ao envelhecimento populacional e à adoção de novos padrões de vida e de consumo instituídos pela sociedade moderna, como por exemplo, as doenças degenerativas e o câncer. Este último configura-se como uma das principais causas de preocupação global, devido às dificuldades encontradas em seu tratamento e os alarmantes dados estatísticos apresentados. Os índices mostram que, somente em 2005, foram registrados 58 milhões de mortes causadas por câncer em todo o mundo, correspondendo a 13% do total de óbitos (WHO, 2007). Nos Estados Unidos, esta enfermidade é a segunda maior causa de morte, perdendo apenas para as doenças cardíacas. Em 2006, foram estimados por volta de 1,4 milhões de novos registros, e mais de 500 mil mortes nesse país (THUN et al.,2006), ao passo que no Brasil calculou-se que, no mesmo ano, existissem 472 mil novos casos. Em 2004, foram registrados 141 mil óbitos, sendo os cânceres mais incidentes os de estômago, pulmão e próstata para homens e mama, pulmão e intestino para mulheres (INCA, 2007a).
Aspectos gerais sobre o câncer
Células cancerosas são aquelas que, devido à ocorrência de uma mutação em seu material genético, passam a ter um crescimento descontrolado; adquirindo inclusive a capacidade de invadir e colonizar espaços reservados para outras células. O acúmulo dessas células anormais em um determinado tecido é denominado tumor ou neoplasma. Esse pode ser considerado benigno, quando a massa celular permanece unida num mesmo tecido, ou maligno, quando essas têm habilidade para escapar do tecido de origem, passando a corrente sanguínea ou linfática, o que leva à formação de tumores secundários ou metástases, em outras partes do organismo (DE VITA JR et al., 1997). Os tumores podem ser classificados de acordo com o tecido e tipo de célula que os originam, por exemplo, de modo geral, sabe-se que os carcinomas são oriundos de células epiteliais, os sarcomas são originários dos tecidos musculares e conjuntivos, enquanto que aqueles que não se enquadram nessas duas categorias, são ditos leucemias, que são derivados do tecido hematopoiético (tecido conjuntivo, presente principalmente na medula óssea, que é especializado na formação de células sangüíneas, como hemácias e leucócitos) e do sistema nervoso. Cada um desses grupos é ainda dividido de acordo com a estrutura do tumor, sua localização e tipo específicos de célula envolvida (DE VITA JR et al., 1997). A maioria dos cânceres parece ser originada a partir de uma única célula que sofreu mutação, originando uma população levemente anormal. Essas células, por sua vez, continuam realizando sucessivos ciclos mutacionais, antes de se tornarem cancerosas. Por isso, o fenômeno de progressão do tumor é lento, podendo levar anos para se concretizar. Desta forma, existem dois genes normais que estão envolvidos na conversão de uma célula normal em uma cancerígena: proto-oncogene e gene supressor de tumor ou antioncogene. Esses genes são responsáveis pela síntese de proteínas importantes para a regulação do crescimento (anti-oncogenes) ou proliferação celular (proto-oncogenes). Logo, há duas possíveis rotas capazes de promover essa diferenciação celular. A primeira delas consiste na hiperestimulação do oncogene, que é originado a partir de uma mutação no alelo normal proto-oncogene, sendo esse um processo dominante, ou seja, deve haver apenas alteração em uma das cópias dos genes. Já a outra possibilidade consiste na inativação ou na deleção do gene inibitório supressor de tumor, sendo esse um processo recessivo; exigindo alterações nas duas cópias dos genes para que ocorra tal mutação (ALBERTS et al., 1994).
Terapias antineoplásicas
Tendo em vista a alta incidência dessa enfermidade em todo o mundo, diversos esforços vêm sendo realizados no intuito de se desenvolver ou aperfeiçoar terapias antineoplásicas. Basicamente, as formas mais utilizadas de combate ao câncer são: a cirurgia, a radioterapia, a hormonioterapia e a quimioterapia. A primeira trata-se da modalidade de tratamento mais antiga e definitiva, que consiste na remoção do tumor, quando este é localizado e encontra-se em condições anatômicas favoráveis. No entanto, em alguns tipos de câncer, as cirurgias não são suficientes, pois as células neoplásicas podem estar disseminadas ou irradiadas em outros tecidos. Assim como a retirada cirúrgica, a radioterapia é indicada, principalmente, para tumores localizados, que não podem ser totalmente removidos ou para aqueles que costumam reincidir após o processo cirúrgico. Essa modalidade consiste na utilização de diferentes radiações ionizantes (Raios X ou Raios gama) que, ao interagirem com os tecidos, aceleram elétrons, promovendo processos químicos como, por exemplo, a ruptura da duplahélice do DNA, que pode causar a morte celular. A grande desvantagem deste método é a existência de efeitos colaterais oriundos da lesão de tecidos normais adjacentes ao tumor. Já a hormonoterapia, por sua vez, consiste na retirada de circulação dos hormônios ao qual o tumor é sensível ou na introdução de substâncias com efeito contrário a esses hormônios no organismo. Esse tipo de tratamento é importante no controle de tumores que se desenvolvem a partir de células dependentes dos hormônios sexuais para crescer, como por exemplo, pode-se citar o câncer da próstata, da mama e do endométrio. A quimioterapia, diferentemente da cirurgia e da radioterapia, é uma forma de tratamento sistêmico à base de fármacos que interferem no processo de crescimento e divisão celular de células cancerígenas. No entanto, a maioria desses fármacos não possui especificidade, logo também são capazes de destruir tecidos sadios, levando a efeitos colaterais como náuseas, vômitos, anemia e mielossupressão. A quimioterapia moderna é baseada na combinação de fármacos (poliquimioterapia), que apresenta resultados mais eficazes que a monoterapia, pois nos tumores há freqüentemente subpopulações de células com sensibilidade diferente às drogas antineoplásicas, além disso, a poliquimioterapia pode diminuir o risco de resistência aos fármacos empregados. Normalmente, a quimioterapia é indicada como tratamento principal em leucemias, linfomas e cânceres de testículo, porém é utilizada geralmente como adjuvante, após cirurgia ou radioterapia, ou como paliativo, em casos graves e avançados da doença, visando melhorar a qualidade de vida do paciente (INCA, 2007b).
A quimioterapia e o ciclo celular
Os agentes quimioterápicos anti-neoplásicos existentes agem tentando regular a ação dos oncogenes e do ciclo celular. Nos mamíferos, o ciclo celular tem duração de aproximadamente 24 horas, sendo dividido em duas fases principais: Mitótica (M) e Interfase (Figura 4). A fase mitótica é a mais crítica, porque envolve a divisão propriamente dita da célula em duas partes (citocinese) e a separação correta dos cromossomos duplicados. No entanto, essa fase é extremamente curta, tendo apenas uma hora de duração. No restante do tempo, a célula encontra-se em interfase, período no qual acontece a preparação de toda a maquinaria celular necessária para que ocorra a fase M. A interfase, por sua vez, é dividida em três estágios S, G1 e G2, dentre os quais se destaca o primeiro deles, pois é quando se processa a duplicação do material genético. As duas outras fases são úteis para que haja tempo suficiente para a célula duplicar sua massa e produzir elementos celulares indispensáveis à divisão. Existe ainda uma fase denominada G0, na qual a célula está em repouso, ou seja, não está em processo de divisão. Na célula tumoral, a fase Go nunca é alcançada, logo o fenômeno de replicação celular é contínuo. Isso acontece, pois nessas células os mecanismos reguladores do ciclo celular sofreram alterações, como por exemplo, sabe-se que, ao perceber algum dano no DNA, os genes supressores de tumor interrompem a multiplicação celular; permitindo que aquele seja reparado; fato que não ocorre em uma célula cancerígena, na qual esse controle é precário ou inexistente (ALBERTS et al., 1994).
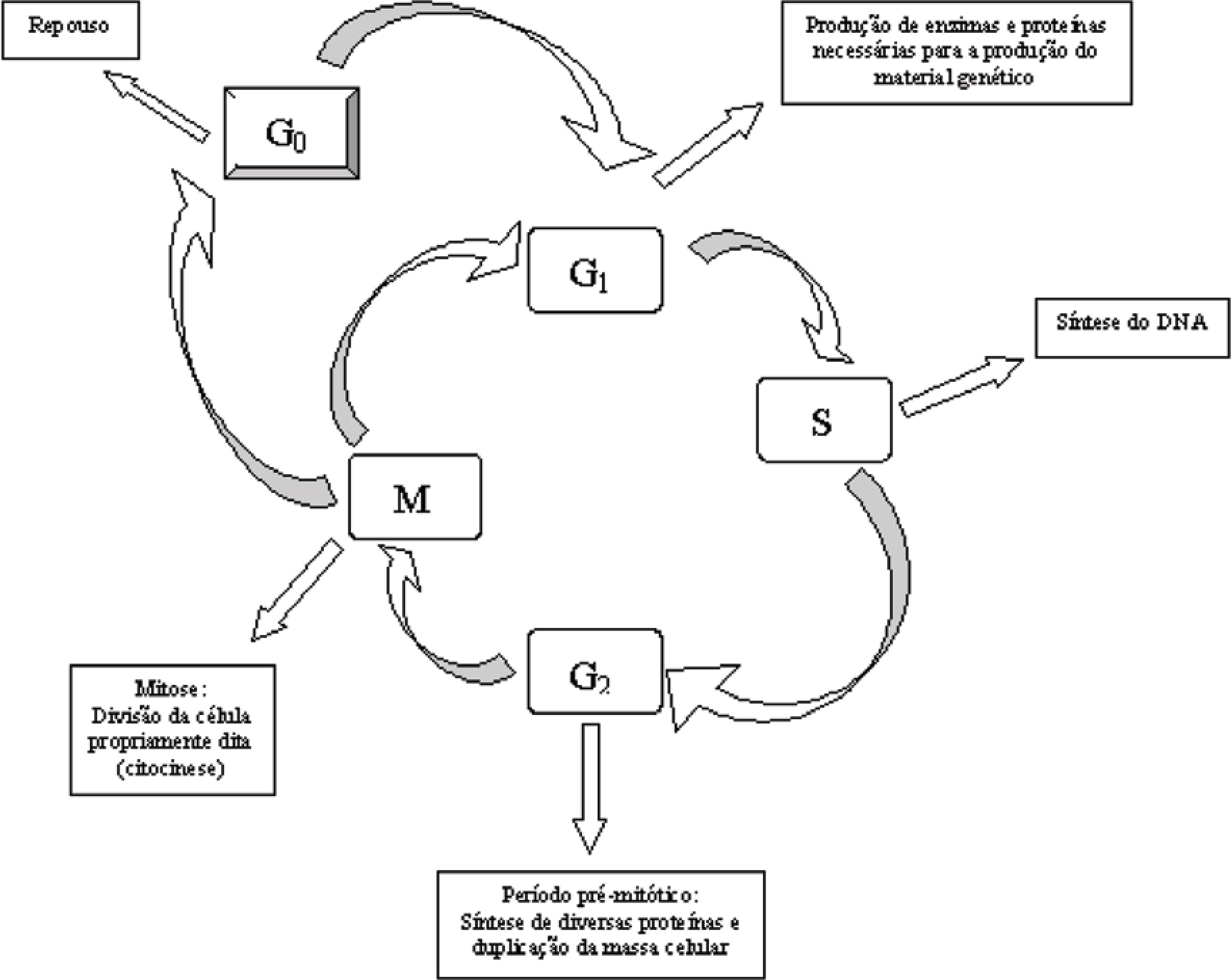
Tendo em vista as fases do ciclo celular, nota-se que os principais agentes quimioterápicos atualmente utilizados atuam em uma ou mais etapas desse processo, impedindo de diferentes maneiras a multiplicação desenfreada das células tumorais. Deste modo, esses fármacos estão divididos em duas grandes categorias: ciclo-inespecíficos e ciclo-específicos. Os primeiros agem nas células que estão ou não se multiplicando, como por exemplo, as mostardas nitrogenadas. Já os agentes cicloespecíficos, como a vincristina, atuam apenas em células que estão em processo de divisão. Na Tabela 1, encontram-se listados as principais classes de fármacos utilizados no combate ao câncer, seus mecanismos de ação gerais e alguns exemplos (CALABRESI; CHABNER, 2001)
| Categorias | Classes | Mecanismos de ação gerais | Exemplos |
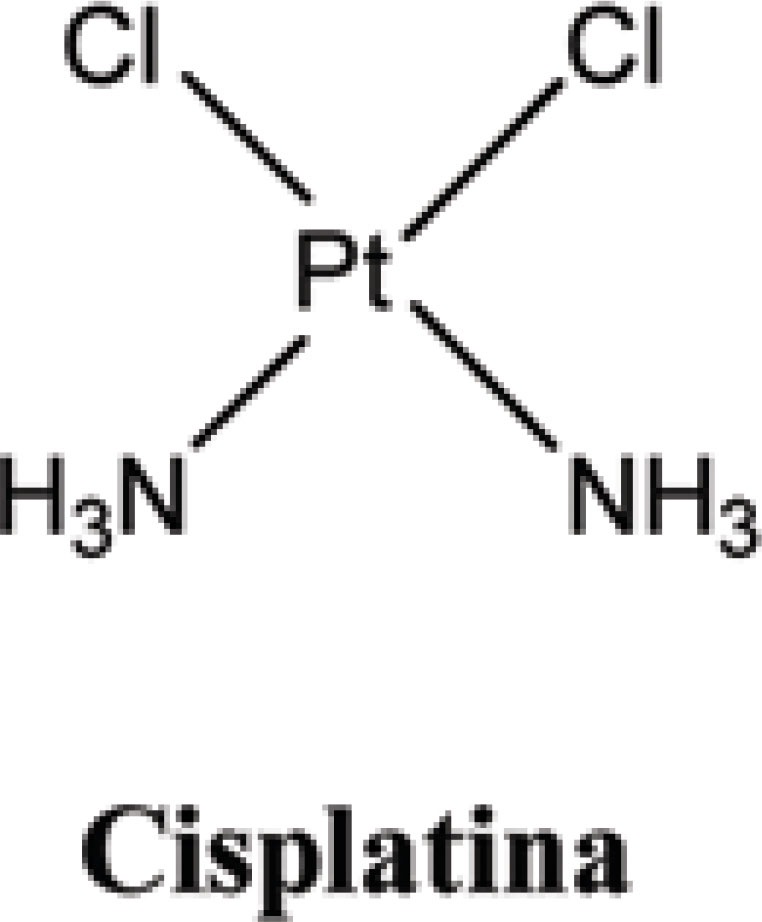
| Cicloinespecíficos | Complexos de coordenação de Platina | Formação de ligações cruzadas intra e interfilamentares no DNA, inibindo a síntese do mesmo e, consequentemente, impedindo o processo de divisão. | Cisplatina, Carboplatina. |
| Antibióticos | Possuem estruturas químicas variáveis, no entanto têm em comum o fato de apresentarem anéis insaturados que permitem a incorporação de elétrons, favorecendo assim a formação de radicais livres reativos que podem se ligar ao DNA e/ou induzirem a apoptose. | Bleomicina, Mitomicina, Antraciclinas, Plicamicina, Dactinomicina. | |
| Agentes alguilantes | Ligam-se a dupla hélice do DNA, impedindo a separação dos dois filamentos, evento esse indispensável para a replicação do material genético e duplicação celular. | Mostardas nitrogenadas, Triazenos, Alquilsulfonatos. | |
| Cicloespecíficos | Agentes antimetabólitos | Inibem a síntese dos componentes essências do DNA e RNA (purinas, ácido fólico e pirimidinas), impedindo a divisão. | Metotrexato (Ácido fólico), Fluorouracil (Pirimidinas), Mercaptopurina e Tioguanina (Purinas). |
| Inibidores mitóticos | Atuam sobre a tubulina, proteína formadora dos microtúbulos, que por sua vez, dão origem ao fuso mitótico. Esse é responsável pela divisão dos cromossomos durante a metáfase. Sendo assim ao bloqueio da divisão celular levando a apoptose. | Vincristina, Vinblastina, Vindesina,Podofilotoxina, Taxol (Paclitaxel) |
Plantas fontes de fármacos anticancerígenos
Dentre as diversas fontes disponíveis na natureza, a utilização de plantas na descoberta de novos compostos orgânicos é certamente a que apresenta a parcela de contribuição mais significativa. A capacidade que as plantas apresentam de desenvolver substâncias essenciais na manutenção da sua sobrevivência frente às adversidades do meio ambiente em que se encontram nos fornece uma vasta variedade de compostos bioativos, em sua maioria de grande complexidade, despertando o interesse de pesquisadores e da indústria farmacêutica na obtenção de novos padrões moleculares. É neste contexto que se pode verificar o importante papel do reino vegetal no desenvolvimento de terapias antineoplásicas, já que muitos dos principais fármacos utilizados na terapêutica atualmente estão incluídos nessa classe como já exemplificados nas FiguraS 2 e 3. Sendo assim, importantes estudos estão sendo descritos na literatura, apresentando outros derivados de plantas como promissores candidatos no desenvolvimento de novas terapias antineoplásicas, onde se destacam alguns em fase de testes clínicos, incluindo: a combretastatina, o flavopiridol, e a bruceantina (Figura 5).
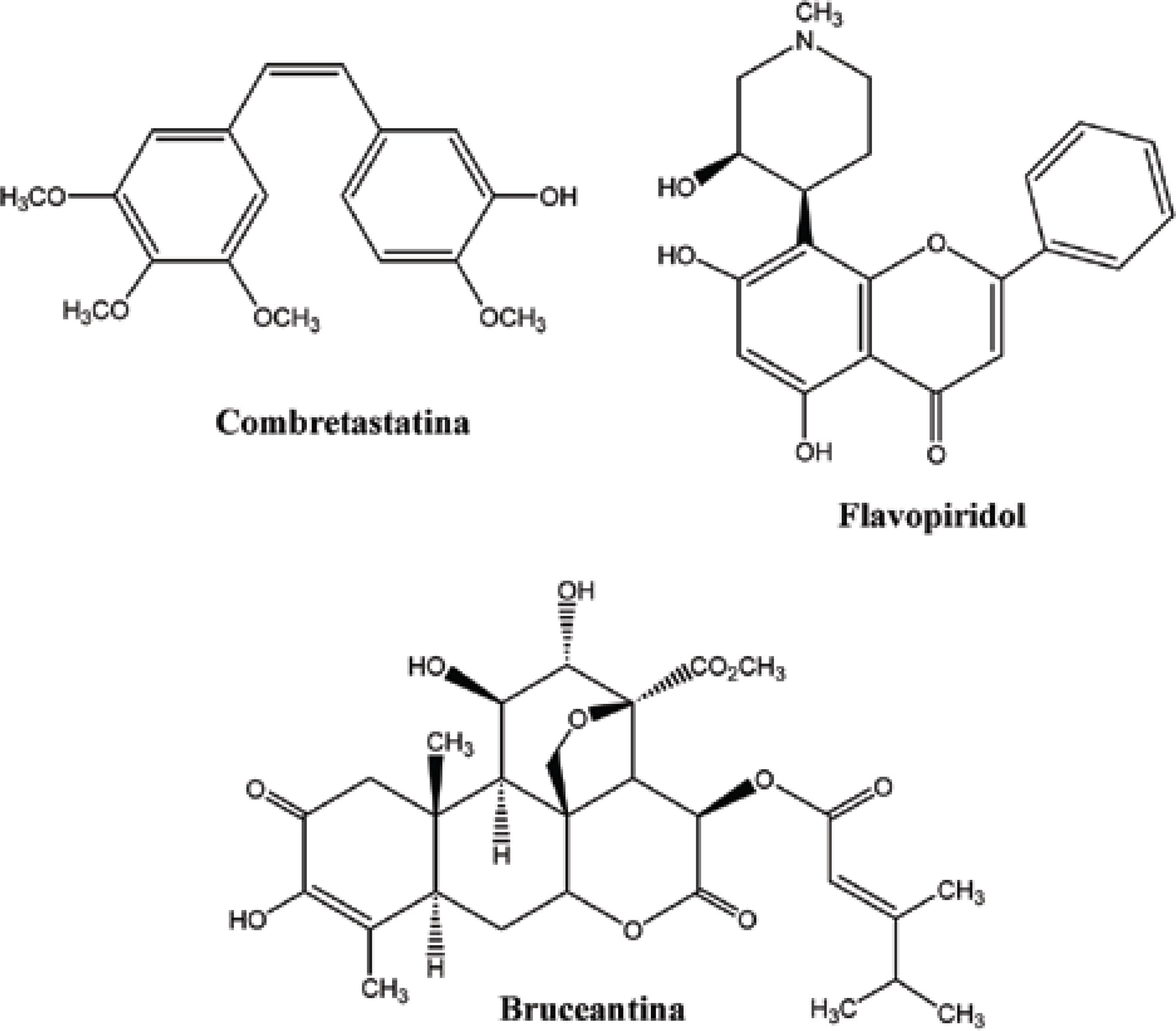
Combretastatina
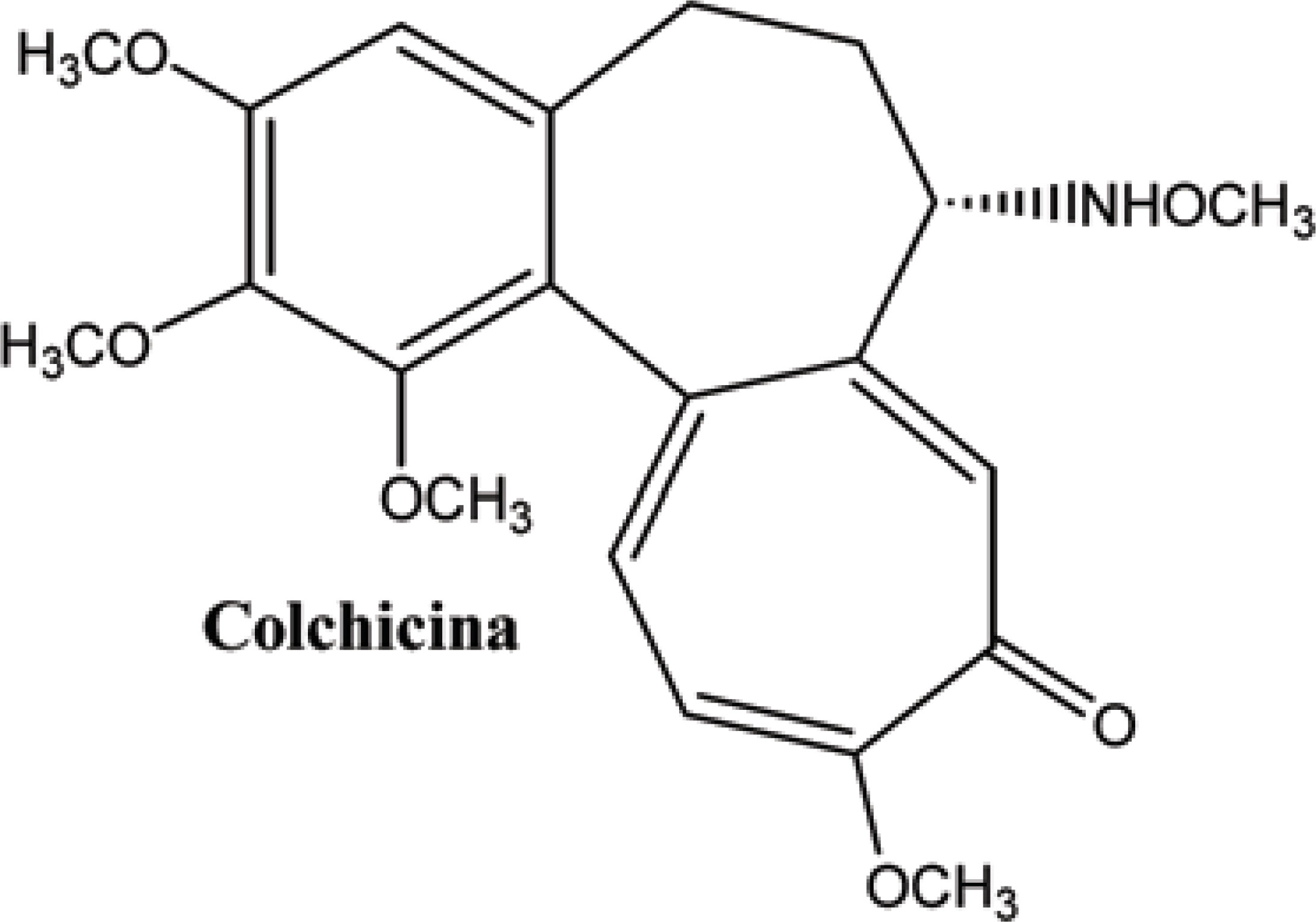
A Combretastatina, derivada dos arbustos do salgueiro africano Combretum caffrum, teve seu mecanismo de ação elucidado pela primeira vez em 1988 por Lin e colaboradores (LIN, 1988). Semelhante ao da colchicina (Figura 6), a combretastatina tem a capacidade de inibir a polimerização da tubulina, o que leva às mudanças morfológicas em células endoteliais, resultando no aumento da permeabilidade vascular tumoral, na redução do fluxo sanguíneo do tumor e, em seguida, na necrose de seu tecido.
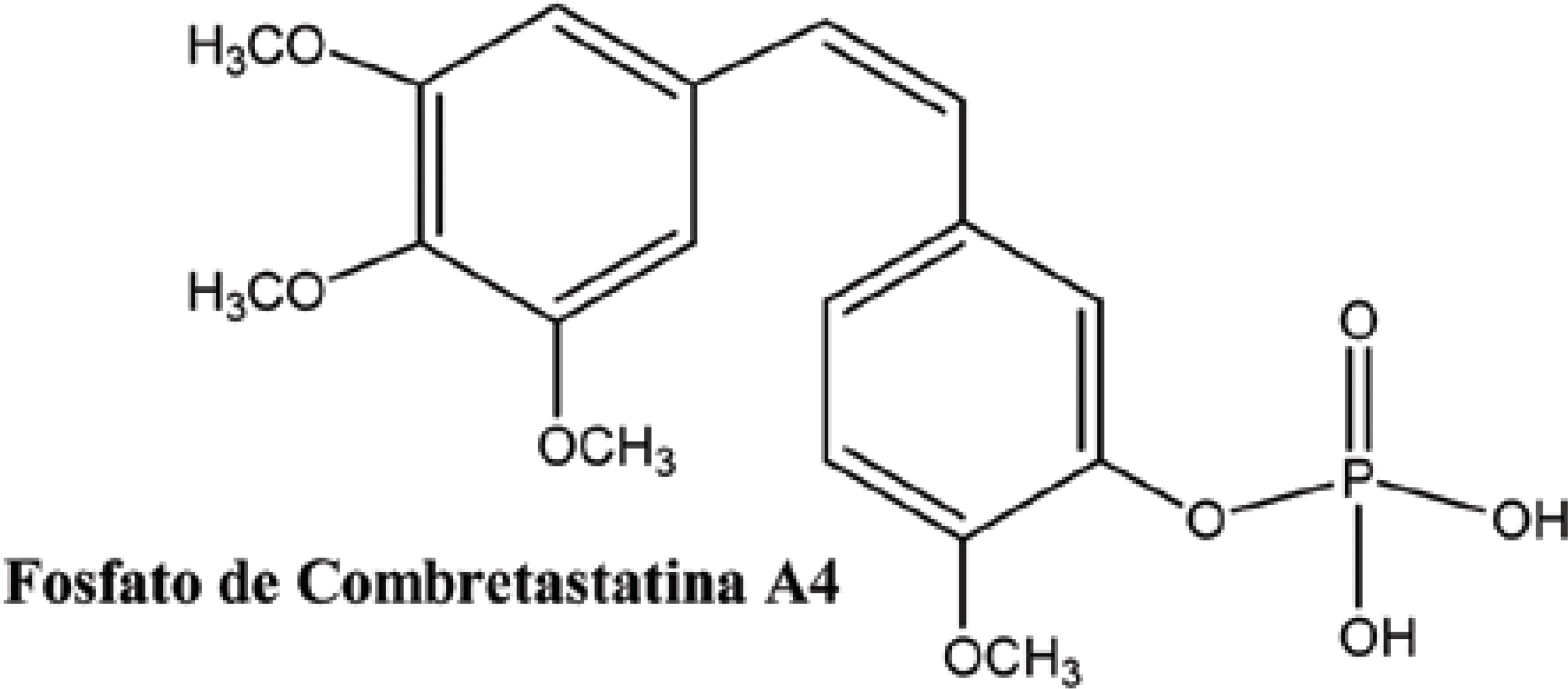
Devido à simplicidade de sua fórmula estrutural, diversos análogos estão sendo sintetizados e testados, 13 destes em fase pré-clínica e três em fase clínica, destacando-se o fosfato de combretastatina A4 (Figura 7), derivados solúveis, que atualmente encontra-se em fase II de triagem clínica.(CRAGG; NEWMAN, 2004).
Flavopiridol
Apresentando resultados promissores, o flavopiridol (Figura 5) é o primeiro inibidor ciclo-dependente da Quinase. Sustância semi-sintética derivada de um alcalóide extraído das folhas e galhos da Amoora Rohituka, seu mecanismo de ação dá-se interferindo na fosforilação de Quinases ciclo-dependentes, dificultando sua ativação e bloqueando o ciclo celular progressivamente até o crescimento na fase G1 ou G2 (SENDEROWICZ, 1999). Alguns estudos em fase II já foram realizados, objetivando verificar a toxicidade do flavopiridol em linfomas de células manto e sua atividade em pacientes com carcinoma em células renais e câncer do cólon (AHN et al., 2007; VELDHUIZEN et al., 2005; KOUROUKIS et al., 2003; KLINDLEN, 2003). Outros estudos vêm sendo realizados em pacientes com tumores sólidos, combinando Flavopiridol com a Cisplatina (Figura 8), Flavopiridol com o Paclitaxel (Figura 2) e os três juntos.
Bruceantina
A bruceantina (Figura 5) é um triterpeno, que foi isolado pela primeira vez em 1972 por Morris e colaboradores, a partir da Brucea antidysenterica, uma árvore encontrada na região da Etiópia (KUPCHAN et al., 1973).Seu principal mecanismo de ação antineoplásica está atribuído à inibição de síntese protéica, que se dá a partir da interferência em sítios da enzima peptidiltrasferase, prevenindo a formação de ligações peptídicas. Alguns estudos com esse fármaco alcançaram a fase II nos testes clínicos, porém, foram interrompidos, por não apresentarem os objetivos desejados. Promissores testes estão sendo iniciados em linhagens de células leucemicas, de linfoma e mieloma. Dados da literatura também demonstram atividade antimalarial da bruceantina (CUENDET; PEZZUTO, 2004; HITOTSUYANAGI et al., 2006).
Fármacos anti-neoplásicos obtidos a partir de microorganismos
Há muitas décadas, sabe-se do potencial dos microorganismos como fonte de novos agentes terapêuticos, pois muitas vezes esses são capazes de produzir metabólitos secundários biologicamente ativos. Assim, desde a descoberta da penicilina, por Fleming, em 1929, muitos outros antibióticos foram isolados de microorganismos diversos, como por exemplo, a neomicina, oriunda do fungo Streptomyces fradiae, obtida em 1949, o cloranfenicol, isolado em 1947 da espécie Streptomyces venezuelae e a kanamicina, obtida em 1957, a partir do fungo Streptomyces kanamyceticus (Figura 6). Outro bom exemplo da utilização dos microorganismos na obtenção de novos fármacos foi à descoberta da lovastatina (Figura 9), um agente redutor do colesterol, isolado primeiramente em 1979, em culturas de Monascus ruber e, posteriormente, em 1974, do fungo Aspergillus terreus, por pesquisadores dos laboratórios Merck. Essa substância trata-se de um inibidor da enzima HMGCo-A Redutase, que está envolvida no metabolismo do colesterol (XIE et al., 2006; NOVAK et al., 1997).
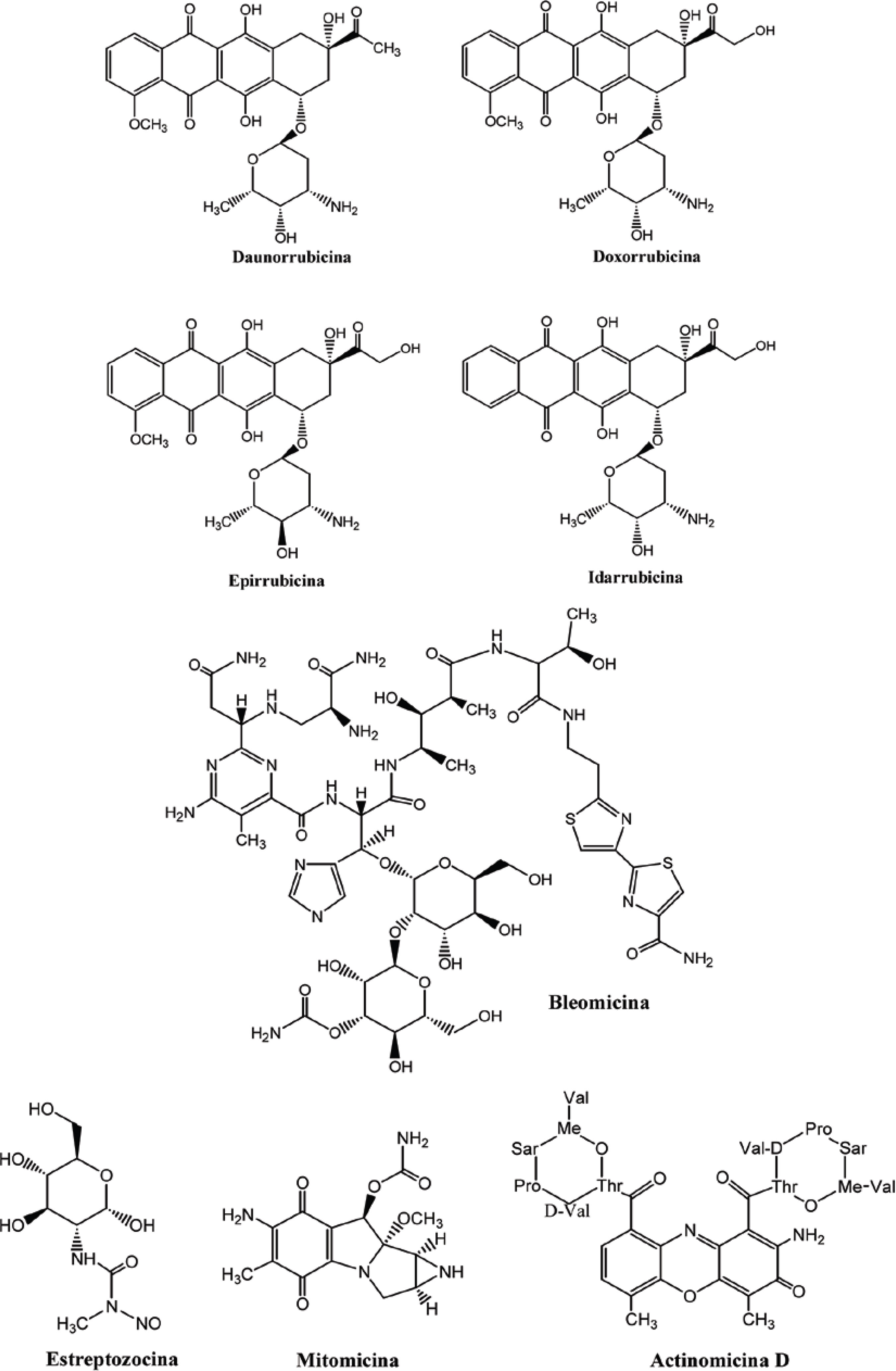
Os microorganismos também se apresentam como importante fonte de agentes antineoplásicos. Atualmente, já são utilizados na terapêutica os seguintes fármacos: daunorrubicina, doxorrubicina, epirrubicina e idarrubicina, substâncias isoladas de cepas de Streptomyces peucetius, na década de 1960 (NERI et al., 2007; TEDESCHI et al., 2007; RITU et al., 2006; TOKARSKA-SCHLATTNER et al., 2006) bleomicina, isolada em 1965, no Japão por Umezawa e colaboradores, obtida a partir de culturas de Streptomyces verticillus (CHLADE-BARTUSIAK et al., 2002). A estreptozocina, obtida da espécie Streptomyces achromogenes (STROSBERG; KVOLS, 2007). Mitomicina, isolada por Wakaki e colaboradores, em 1958, da espécie Streptomyces caespitosus (CHLADE-BARTUSIAK et al., 2002) e actinomicina D, isolada de Streptomyces parvallus ( SPANO et al., 2007) (Figura 10).
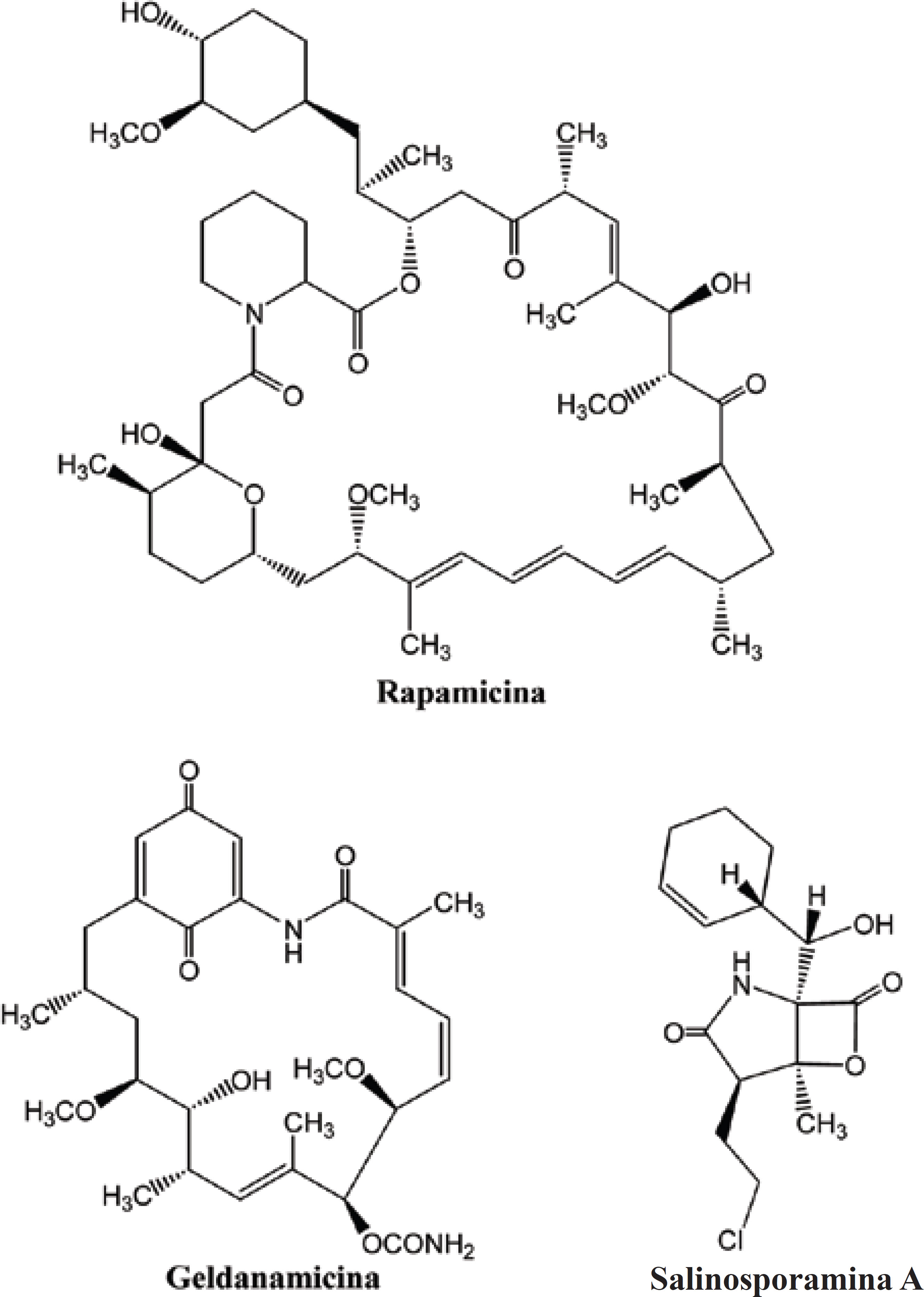
Mesmo existindo uma grande quantidade de antibióticos já utilizados no tratamento do câncer, esses continuam sendo alvos de estudos como fonte de novos agentes anti-neoplásicos. Como exemplos, podemos citar três novas substâncias, que atualmente encontram-se em fase de testes clínicos: Rapamicina, Geldanamicina e Salinosporamida A (Figura 11).
Rapamicina
A Rapamicina é um peptídeo isolado em 1975, a partir do microorganismo Streptomyces hygroscopicus. Essa substância possui atividade imunossupressora, já aprovada pelo FDA em setembro de 1999, sendo indicada para prevenção da rejeição de órgãos transplantados. Devido as suas propriedades citotóxicas, a Rapamicina encontra-se atualmente em fase II de testes clínicos. O seu mecanismo de ação consiste em promover a apoptose, por meio da ligação do fármaco a um receptor intracelular, chamado FKBP (FKB 506 Binding Protein).
Esse complexo se liga a um regulador da fase G1 do ciclo celular, chamado FRAP, fazendo com que a transição G1-S seja interronpida, fato esse que induz instantaneamente a apoptose (ZANGARI et al., 2006; SCHULTE; NECKERS, 1998; ALBERTS et al., 1993).
Geldanamicina (GA)
A geldanamicina (GA) também é uma substância isolada da espécie Streptomyces hygroscopicus, mas seu mecanismo de ação antineoplásico é bastante diferente daquele descrito acima. Esse fármaco se liga a um tipo especial de proteína chamada HSP (Heat Shock Protein), produzida por todas as células, sendo responsáveis pelas respostas a situações de estresse, como calor, exposição a compostos tóxicos e outras. A GA se liga especificamente a HSP 90, que atua como uma chaperona molecular, que é uma substância presente no interior das células, responsável pela ligação e estabilização de outras proteínas que promovem o transporte por meio das membranas ou degradação de outras moléculas. A HSP 90 está envolvida no processo de progressão do câncer. Logo quando ocorre a ligação com a GA, essa proteína perde sua habilidade de atuar como chaperona, tornando-se incapaz de auxiliar a célula a degradar as proteínas envolvidas na progressão do tumor. Atualmente, essa substância encontra-se em fase I/II de testes clínicos (SOLAR et al., 2007; BAGATELL et al., 2005; BEDIN et al., 2004).
Salinosporamida A (NPI-0052)
A salinosporamida A (NPI-0052) é obtida da bactéria Salinispora tropica, que é encontrada em sedimentos de oceanos tropicais. Essa substância encontra-se em fase I de testes clínicos e possui um mecanismo de ação bastante promissor, que consite na inibição do proteossomo. Esse importante constituinte celular é responsável pela degradação de proteínas normais que já foram utilizadas ou foram metabolizadas erroneamente, sendo o acúmulo dessas no citosol extremamente prejudicial ao bom funcionamento da célula, induzindo a apoptose. Os proteossomos atuam juntamente com a molécula de ubiqüitina, que encontra proteínas-alvo a serem destruídas e as endereça para o centro do proteossomo. A salinosporamida A apresenta atividade frente a uma grande variedade de linhagens de células cancerígenas como mielomas, leucemias, próstata e ademais vem revelando pouca toxidade às células normais (CAUBERT et al., 2007; VENKAT et al., 2005; ROBERT et al., 2003).
Os produtos naturais marinhos e sua importância no combate ao câncer
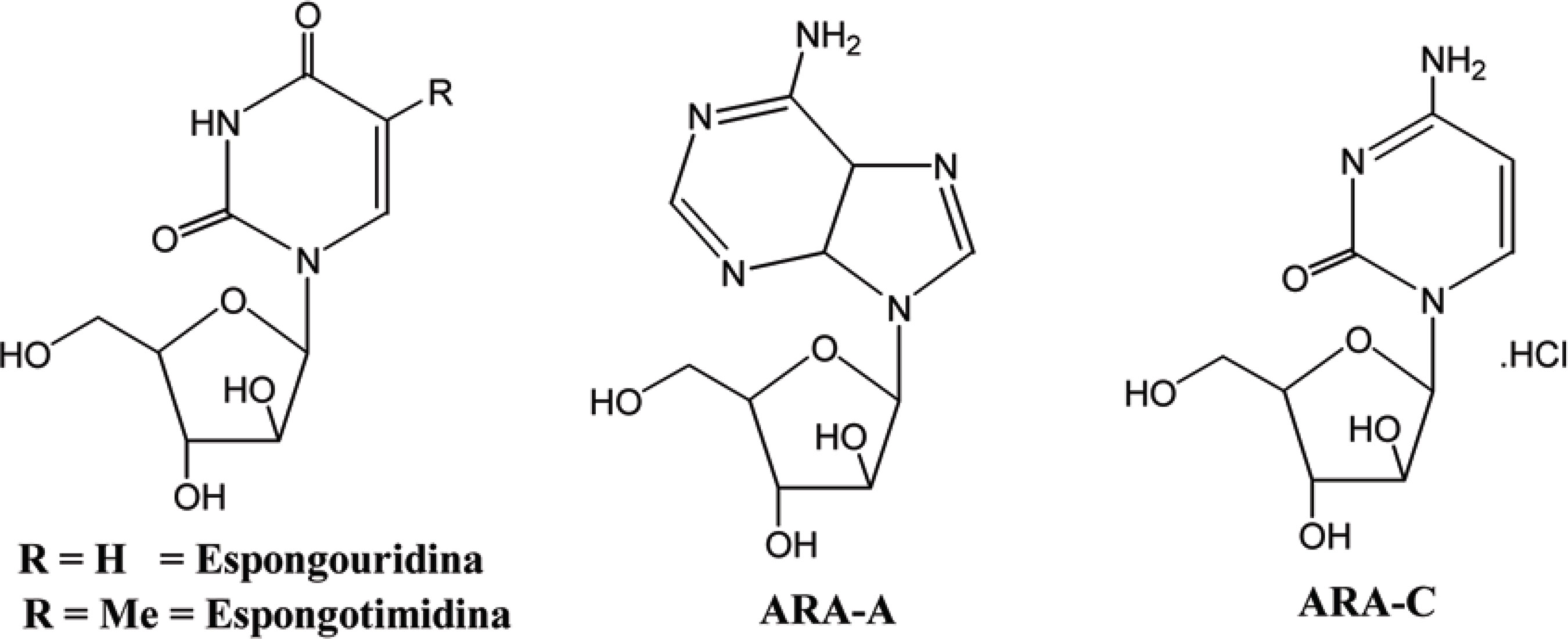
Em todo o mundo, o ambiente marinho vem despertando interesse como potencial fonte de descoberta de novos fármacos, devido a sua enorme biodiversidade e vasta extensão, que ainda são pouco explorados nos dias de hoje. Milhões de organismos marinhos produzem substâncias que são utilizadas na comunicação, defesa, predação, inibição do desenvolvimento de competidores, reprodução; ou simplesmente como produto de seu metabolismo. Sendo assim, essas moléculas podem apresentar atividades biológicas diversas, sendo úteis no desenvolvimento de novas terapias contra muitas enfermidades (DONIA; HAMANN, 2003). Um bom exemplo da utilização das substâncias produzidas por seres marinhos no desenvolvimento de novos fármacos consistiu no isolamento dos nucleosídeos espongouridina e espongotimidina por Bergmann e colaboradores, em 1955 (BERGMANN; BURKE, 1955). Esses foram isolados da esponja Tethya crypta e serviram de modelo para a descoberta de moléculas como ARA-A e ARA-C, que são atualmente utilizados como antiviral e antineoplásico no tratamento de leucemia, respectivamente (Figura 12). A Tabela 2 ilustra o potencial das substâncias isoladas de organismos marinhos, já utilizadas como agentes terapêuticos com diferentes finalidades. Na Figura 13 encontram-se representadas as estruturas das substâncias contidas na tabela mencionada a seguir.
| Fármaco | Origem | Atividade | Ano de comercialização |
| Cefalosporina C | Cephalosporium sp (Fungo marinho) | Antibiótico | 1965 |
| Zidovudina (AZT) | Tethya crypta (Esponja) | Antiviral (inibidor da transcriptase reversa) | 1987 |
| Ácido Kaínico | Digenea simplex (Alga vermelha) | Anti-helmíntico Inseticida | Início dos anos 90 |
| Ziconotida | Conus magnus (Molusco) | Analgésico | 1999 |
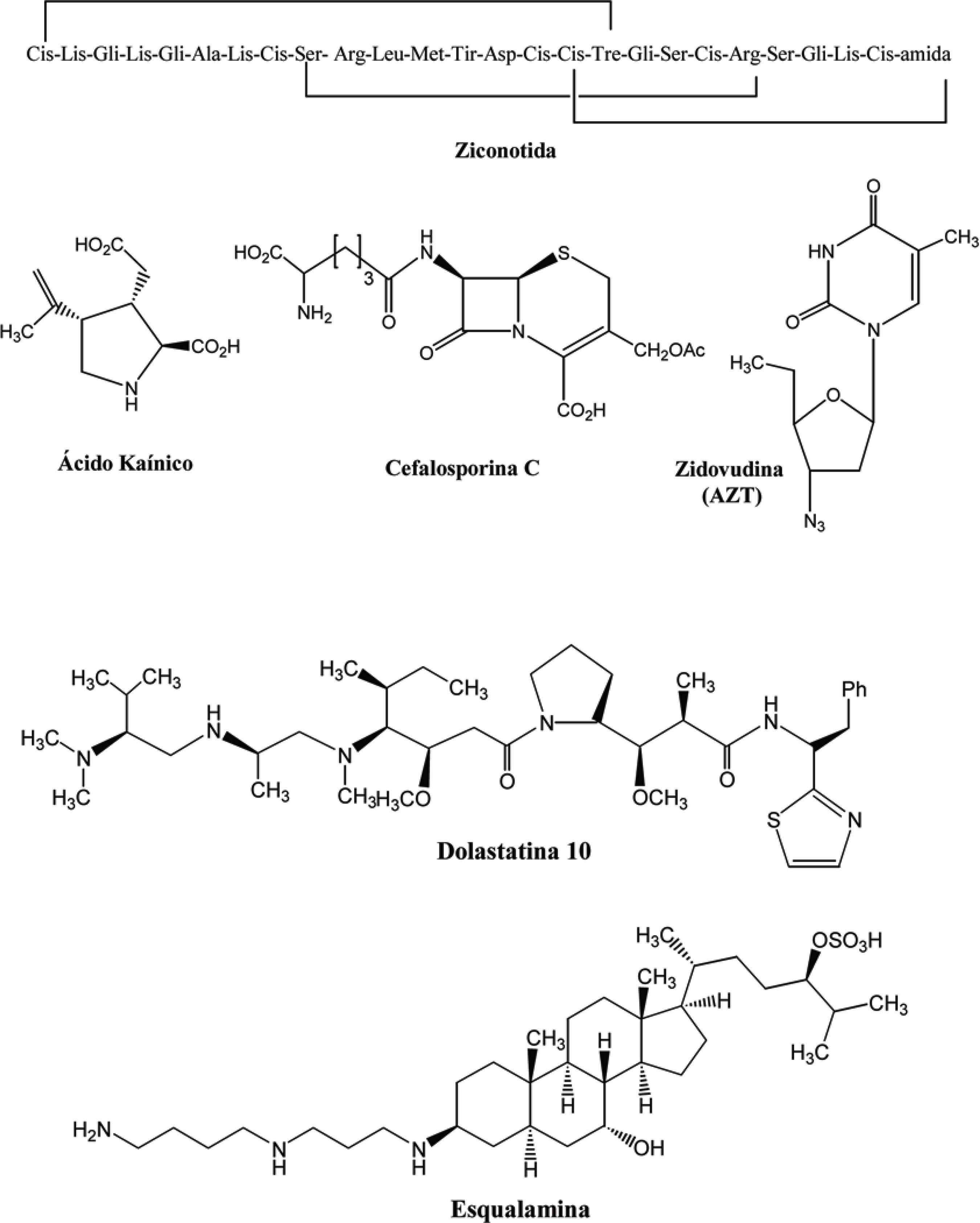
Tendo em vista a alta capacidade de aproveitamento dos produtos naturais marinhos como fonte de novos fármacos, muitas substâncias têm sido isoladas e testadas farmacologicamente contra o câncer. Na Figura 14, encontram-se algumas moléculas com potencial antineoplásico, que atualmente estão em fase II de testes clínicos.
Dolastatina 10
O molusco Dolabella auricularia, oriundo do oceano Índico, apresentou-se, já na década de 1970 como um potente agente antitumoral, descoberto por Pettit e colaboradores. Entretanto, devido às pequenas quantidades de princípio ativo isoladas, somente quinze anos depois, é que a dolastatina 10 teve a sua estrutura elucidada. Seu mecanismo de ação consiste na inibição da proliferação celular e indução do apoptose, por meio da interação com a tubulina, resultando em alterações da função dos microtúbulos. Há sinergismo na atividade antitumoral quando é ministrado em combinação com a Bryostatina-1 ou com os alcalóides da vinca (ALCARO et al., 2007; MADDEN et al., 2005).
Esqualamina
A Esqualamina é um aminoesterol isolado do tubarão Squalus acanthias, proveniente da costa inglesa, por Zasloff e colaboradores em 1993, tendo sido posteriormente licenciado a Maganin Pharmaceutical para realização dos testes de fase II. Essa substância inibe a angiogênese (processo pelo qual, novos vasos sangüíneos são formados) e o crescimento de tumores sólidos (CHEN et al., 2006; CHO, KIM; 2002).
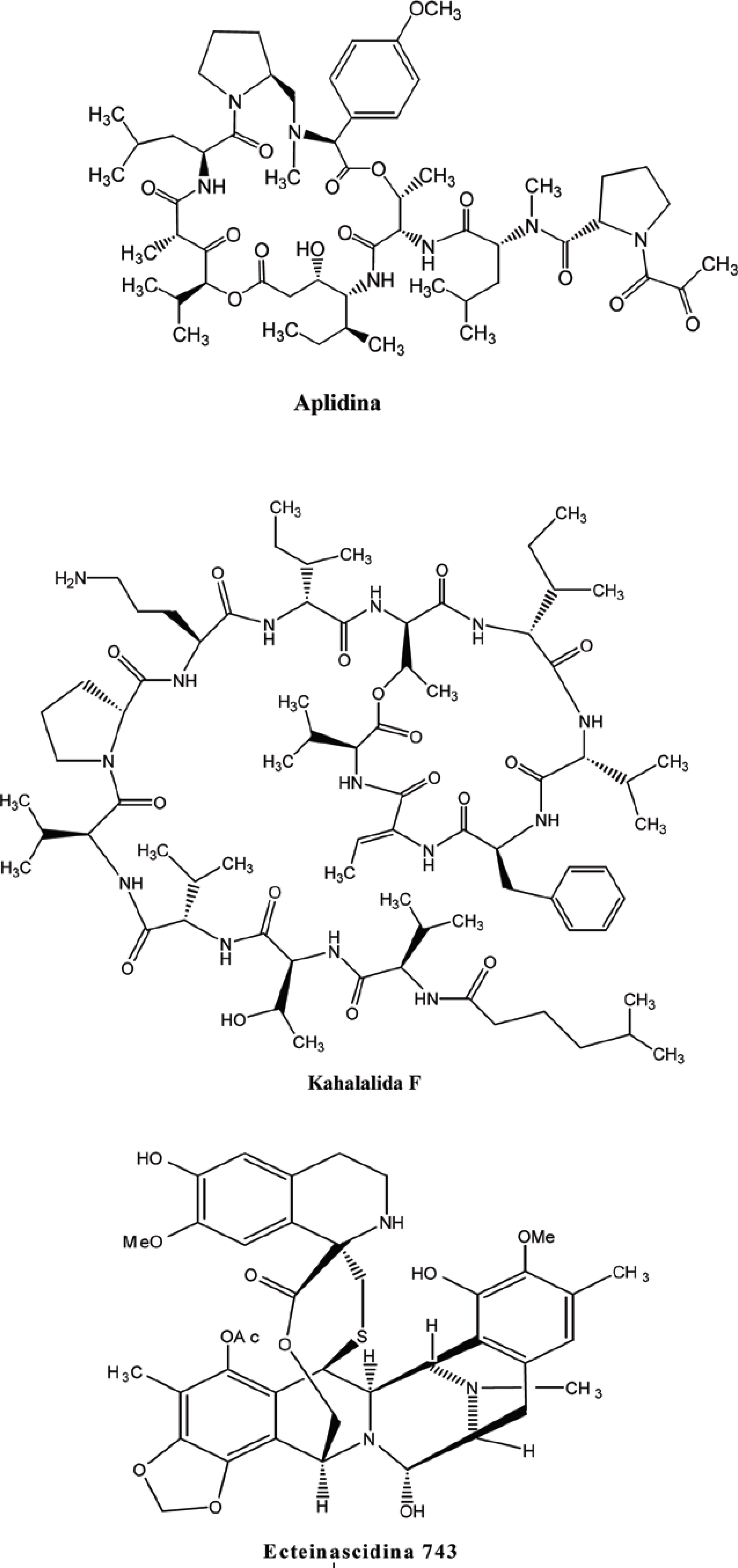
Aplidina
Substância isolada da espécie mediterrânea Aplidium albicans, a aplidina possui um mecanismo de ação único, que consiste na inibição da enzima ornitina descarboxilase, fundamental para o processo de crescimento do tumor e para angiogênese (BEUMER et al., 2007; GARCÍA-FERNÁNDEZ et al., 2002; ERBA et al., 2002). Os efeitos anti-angiogênicos são aumentados pela diminuição da secreção de fator de crescimento do endotélio vascular e redução da expressão de receptores para o fator acima citado.
Kahalalida F
A substância kahalalida F foi isolada, primeiramente, no Hawai a partir do molusco Elysia rufescens e também da macroalga Bryopsis sp, da qual esse animal se alimenta. No entanto, a concentração de KF no molusco é muito mais significativa do que na referida alga. Nos anos 90, essa molécula foi licenciada a PharmaMar pela Universidade do Hawai, mas apenas no ano 2000, foram iniciados os primeiros testes clínicos de fase I, frente ao câncer de próstata independente de andrógeno. O mecanismo de ação deste fármaco ainda não é totalmente conhecido, todavia sabe-se que seus alvos intracelulares são os lisossomos, que têm suas membranas despolarizadas, o que gera a indução da morte celular por necrose. Por ter como alvo essas organelas, o KF tem apresentado resultados promissores no tratamento de tumores com alta atividade lisossomal, como o câncer de próstata (GRACIA et al., 2006; RADEMAKER-LAKHAI et al., 2005).
Ecteinascidina 743 (ET-743)
A ecteinascidina 743 (ET-743) é um alcalóide tetraisoquinolínico extraído de Ectenaiscidia turbinata, habitante do mar do Caribe, cuja atividade tumoral já havia sido reportada desde 1969, por Sigel e colaboradores. No entanto, somente nos anos 90 houve a elucidação estrutural do composto, por dois grupos de pesquisa (Rinehart, e Wright e colaboradores) simultaneamente. Possui alta atividade citotóxica e atua alterando a interação do DNA com fatores de transcrição e outras proteínas. Além disso, gera um atraso na progressão do ciclo celular de G1 para G2, que inibe a síntese do DNA, fazendo com que o ciclo estacione em G2. Isso ocasionalmente leva à indução do apoptose independente da proteína p53. ET-743 também interage com outros alvos moleculares, como por exemplo, no sistema de microtúbulos (ERBA et al., 2001; MINUZZO et al., 2000). Esse fármaco mostrou-se ativo contra melanomas e carcinomas de pulmão, cérebro, ovário e mama (BEUMER et al., 2007; CHEN et al., 2006; LE CESNE et al., 2005; GARCIA et al., 2004).
Briostatina-1
A briostatina-1 foi isolada por Pettit e colaboradores, em 1982, da espécie Bugula neritine, encontrado na costa oeste da Flórida. Desde então, testes clínicos têm sido realizados, constatando-se que o uso desta substância isoladamente no tratamento de diferentes neoplasias não é adequado, sugerindo a combinação com outros agentes terapêuticos como vincristina, vimblastina, paclitaxel, cisplatina e ARA-C. Esses estudos têm alcançado promissores resultados, visto que a briostatina-1 tem um mecanismo de ação peculiar, pois se trata de um potente ativador da proteína quinase C. Além de ter efeito antagonista sobre os forbol-ésteres promotores de tumor, a briostatina-1, induz ainda a diferenciação de células mielóides e linfóides e promove agregação plaquetária e hematopoiese. Essa substância parece ainda modular a expressão dos genes bcl-2 e p53, induzindo a apoptose (KECK et al., 2006; SUN; ALKON, 2006; BAI et al., 2002; GSCHWEND, 2000; VARTERASIAN et al., 1998).
Conclusão
Atualmente, a importância dos produtos naturais na luta contra o câncer pode ser comprovada pelo grande número de medicamentos no mercado de origem natural ou semi-sintética que salvam a vida de milhares de pessoas. Devido à sua importância, os produtos naturais extraídos de diversas fontes têm sido amplamente estudados por diferentes grupos de pesquisas e indústrias farmacêuticas, na promessa de novas perspectivas para a obtenção de agentes quimioterápicos inéditos e mais potentes, seletivos e com menores efeitos colaterais.
Referências
AHN, Y. M.; VOGETI, L.; LIU, C.; SANTHAPURAM, H. K. R.; WHITE, J. M.; VASANDANI, V.; MITSCHER, L. A.; LUSHINGTON, G. H.; HANSON, P. R.; POWELL, D. R.; HIMES, R. H.; ROBY, K. F.; YE, Q.; GEORG, G. Design, synthesis, and antiproliferative and CDK2-cyclin a inhibitory activity of novel flavopiridol analogues. Bioorganic & Medicinal Chemistry, v.15, n.2, p.702-713, 2007.
ALBERTS, B.; BRAY, D.; LEWIS, J.; RAFF, M.; ROBERTS, K.; WATSON, J. D. Molecular biology of the cell, Garland Publishing; New York & London; 1994.
ALBERTS, M. W.; WILLIAMS, R.T.; BROWN, E. J.; TANAKA, A.; HALL, F. L.; SCHREIBER, S. L. FKBP-rapamycin inhibits a cyclin-dependent kinase activity and a cyclin D1-Cdk association in early G1 of an osteosarcoma cell line. Journal of Biological chemistry, v.268, n.30, p.22825-22829, 1993.
ALCARO, S.; MARINO, T.; ORTUSO, F.; RUSSO, N. Conformational behavior of antineoplastic peptides Dolastatin 10 and Dolastatin 15 from Monte Carlo and molecular dynamics simulations. International Journal of Quantum Chemistry, v.107, n.2, p.318-325, 2007.
BAI, X.C.; LIU, AN-LING, LUO, SHEN-QIU. Phospholipase C is required for survival in heat stress: involvement of protein kinase C-dependent Bcl-2 phosphorylation. Journal of Biochemistry, v.131, n.2, p.207-212, 2002.
BAGATELL, R.; BELIAKOFF, J.; DAVID, C. L.; MARRON, M. T.; WHITESELL, L. Hsp90 inhibitors deplete key anti-apoptotic proteins in pediatric solid tumor cells and demonstrate synergistic anticancer activity with cisplatin. International Journal of Cancer, v.113, n.2, p.179-188, 2005.
BEDIN, M.; GABEN, A. M.; SAUCIER, C.; MESTER, J. Geldanamycin, an inhibitor of the chaperone activity of HSP90, induces MAPK-independent cell cycle arrest. International Journal of Cancer, v.109, n.5, p.643-652, 2004.
BERGMANN, W.; BURKE, D. C. Contributions to the study of marine products. The nucleosides of sponges. Journal of Organic Chemistry, v.20, n.11, p.1501-1507, 1955.
BEUMER, J. H.; RADEMAKER-LAKHAI, J. M.; ROSING, H.; HILLEBRAND, M. J. X.; BOSCH, T. M.; LAZARO, L. L.; SCHELLENS, J. H. M.; BEIJNEN, J. H. Metabolism of trabectedin (ET-743, Yondelis™) in patients with advanced cancer. Cancer Chemotherapy and Pharmacology, v.59, n.6, p.825-837, 2007.
BRANDON, E. F. A.; SPARIDANS, R. W.; OOIJEN, R. D.; MEIJERMAN, I.; LAZARO, L. L.; MANZANARES, I.; BEIJNEN, J. H.; SCHELLENS, J. H. M. In vitro characterization of the human biotransformation pathways of aplidine, a novel marine anticancer drug. Investigational New Drugs, v.25, n.1, p.9-19, 2007.
CHEN, X.; CHEN, J.; ZHU, J. Synthetic studies on ecteinascidin 743 (Et 743): asymmetric synthesis of a highly oxygenated tetrahydroisoquinoline via a key phenolic Mannich reaction. Synthesis, n.23, p.4081-4086, 2006.
CALABRESI, P.; CHABNER, B. A. Chemotherapy of neoplasic diseases, Em Goodman e Gilman´s: the pharmacological basis of therapeutical, Mac-Graw-Hill; New York; 2001.
CAUBERT, V.; MASSE, J.; RETAILLEAU, P.; LANGLOIS, N. Stereoselective formal synthesis of the potent proteasome inhibitor: salinosporamide A. Tetrahedron Letters, v.48, n.3, p.381-384, 2007.
CHEN, W.; SHAO, X.; MOELLERING, R.; WENNERSTEN, C.; REGEN, S. L.; Bioconjugate Chemistry, v.17, n.6, p.1582-1591, 2006.
CHLADE-BARTUSIAK, K.; STEMBALSKA-KOZLOWSKA, A.; BERNADY, M.; KUDYBA, M.; SASIADEK, M. Analysis of adaptive response to bleomycin and mitomycin. Mutation Research, Genetic Toxicology and Environmental Mutagenesis, v.513, n.1, p.75-81, 2002.
CHO, J. J.; KIM, Y. T. Sharks: A Potential Source of Antiangiogenic Factors and Tumor Treatments. Marine Biotechnology, v.4, n.6, p.521-525, 2002.
CRAGG, G. M.; NEWMAN, D. J. A Tale of Two Tumor Targets: Topoisomerase I and Tubulin. The Wall and Wani Contribution to Cancer Chemotherapy. Journal of Natural Products, v.67, n.2, p.232-244, 2004.
CUENDET, M.; PEZZUTO, J. M. Antitumor Activity of Bruceantin: An Old Drug with New Promise. Journal of Natural Products, v.67, n.2, p.269-272, 2004.
DE VITA JR., V. T.; HELLMAN, S.; ROSENBERG, S. A. Cancer: Principles of Oncology, Lippincott-Raven Publishers; Phifadelphia, 1997.
DONIA, M.; HAMANN, M. T. Marine natural products and their potential applications as anti-infective agents. Lancet Infectious Diseases, v.3, n.6, p.338-348, 2003.
ERBA, E.; BASSANO, L.; DI LIBERTI, G. Cell cycle phase perturbations and apoptosis in tumour cells induced by aplidine. British Journal of Cancer, v.86, n.9, p.1510-1517, 2002.
ERBA, E.; BERGAMASCHI, D.; BASSANO, L. Ecteinascidin-743 (ET-743), a natural marine compound, with a unique mechanism of action. European Journal of Cancer, v.37, n.1, p.97-105, 2001.
GARCÍA-FERNÁNDEZ, L.F.; LOSADA, A.; ALCALDE, V. Aplidin induces the mitochondrial apoptotic pathway via oxidative stress-mediated JNK and p38 activation and protein kinase C. Oncogene, v.21, n.49, p.7533-7544, 2002.
GARCIA, C. R.; SUPKO, J.G.; MANOLA, J. Phase II and Pharmacokinetic Study of Ecteinascidin 743 in Patients With Progressive Sarcomas of Soft Tissues Refractory to Chemotherapy. Journal of Clinical Oncology, v.22, n.8, p.1480-1490, 2004.
GRACIA, C.; ISIDRO-LLOBET, A.; CRUZ, L. J.; ACOSTA, G. A.; ALVAREZ, M.; CUEVAS, C.; GIRALT, E.; ALBERICIO, F. Convergent Approaches for the Synthesis of the Antitumoral Peptide, Kahalalide F. Study of Orthogonal Protecting Groups. Journal of Organic Chemistry, v.71, n.19, p.7196-7204, 2006.
GSCHWEND, J.E. Bryostatin 1 induces prolonged activation of extracellular regulated protein kinases in and apoptosis of LNCaP human prostate cancer cells overexpressing protein kinase C. Molecular Pharmacology, v.57, n.6, p.1224-1234, 2000.
HITOTSUYANAGI, Y.; KIM, I. H.; HASUDA, T.; YAMAUCHI, Y.; TAKEYA, K. A structure-activity relationship study of brusatol, an antitumor quassinoid. Tetrahedron, v.62, n.17, p.4262-4271, 2006.
INCA: Instituto Nacional de Câncer. http://www.inca.gov.br/situacao/arquivos/ocorrencia_magnitude_cancerbrasil.pdf; Acessado em 15/04/2007a.
INCA: Instituto Nacional de Câncer. http://www.inca.gov.br/conteudo_view.asp?id=101, Acessado em 15/04/2007b.
JULSING, M. K.; KOULMAN, A.; WOERDENBAG, H. J.; QUAX, W. J.; KAYSER, O. Combinatorial biosynthesis of medicinal plant secondary metabolites. Biomolecular Engineering, v.23, n.6, p.265-279, 2006.
KECK, G. E.; WELCH, D. S.; POUDEL, Y. B. Synthetic studies toward bryostatin 1: preparation of a C1-C16 fragment by pyran annulation. Tetrahedron Letters, v.47, n.47, p.8267-8270, 2006.
KLINDLEN, H. L. Phase II study of flavopiridol in patients with advanced colorectal cancer. Annals of Oncology, v.14, p.1270-1273, 2003.
KOUROUKIS, C. T.; BELCH, A.; CRUMP, M.; EISENHAUER, E.; GASCOYNE, R. D.; MEYER, R.; LOHMANN, R.; LOPEZ, P.; POWERS, J.; TURNER, R.; CONNORS, J. M. Flavopiridol in Untreated or Relapsed Mantle-Cell Lymphoma: Results of a Phase II Study of the National Cancer Institute of Canada Clinical Trials Group. Journal of Clinical Oncology, v.21, n.9, p.1740-1745, 2003.
KUPCHAN, S. M.; BRITTON, R. W.; ZIEGLER, M. F.; SIGEL, C. W. Bruceantin, a new potent antileukemic simaroubolide from Brucea antidysenterica. Journal of Organic Chemistry,v.38, n.1, p.178-179, 1973.
LE CESNE, L.; BLAY, J.Y.; JUDSON, I. Phase II Study of ET-743 in Advanced Soft Tissue Sarcomas: A European Organization for the Research and Treatment of Cancer (EORTC) Soft Tissue and Bone Sarcoma Group Trial. Journal of Clinical Oncology, v.23, n.3, p.576-584, 2005.
LIN, C. M. Interactions of tubulin with potent natural and synthetic analogs of the antimitotic agent combretastatin: a structure-activity study. Molecular Pharmacology, v.34, n.2, p.200-208, 1988.
MADDEN, T.; TRAN, H. T.; BECK, D.; HUIE, R.; NEWMAN, R. A.; PUSZTAI, L.; WRIGHT, J. J.; ABBRUZZESE, J. L. Novel Marine-derived Anticancer Agents: A Phase I Clinical, Pharmacological, and Pharmacodynamic Study of Dolastatin 10 (NSC 376128) in Patients with Advanced Solid Tumors. Clinical Cancer Research, v.6, n.4, p.1293-1301, 2000.
MANN, J. Natural products in cancer chemotherapy: past, present and future. Nature Reviews Cancer, v.2, n.2, 143-148, 2002.
MINUZZO, M.; MARCHINI, S.; BROGGINI, M.; FAIRCLOTH, G.T.; D’INCALCI, M.; MANTOVANI, R. Interference of transcriptional activation by the antineoplastic drug ecteinascidin-743. Proceedings of the National Academy of Sciences of the United States of America, v.97, n.12, p. 6780-6784, 2000.
NERI, B.; PANTALEO, P.; GIOMMONI, E.; GRIFONI, R.; PAOLETTI, C.; ROTELLA, V.; PANTALONE, D.; TADDEI, A.; MERCATELLI, A.; TONELLI, P. Oxaliplatin, 5-fluorouracil/leucovorin and epirubicin as first-line treatment in advanced gastric carcinoma: a phase II study. British Journal of Cancer, v.96, p.1043-1046, 2007.
NEWMAN, D. J.; CRAGG, G. M.; SNADER, K. M. Natural Products as Sources of New Drugs over the Period 1981-2002. Journal of Natural Products, v.66, n.7, p.1022-1037, 2003.
NOVAK, N.; GERDIN, S.; BEROVIC, M. Increased lovastatin formation by Aspergillus terreus using repeated fed-batch process. Biotechnology Letters, v.19, n.10, p.947-948, 1997.
POMMIER, Y. Topoisomerase I inhibitors: camptothecins and beyond. Nature Reviews Cancer, v.6, n.10, p.789-802, 2006.
RADEMAKER-LAKHAI, J.; HORENBLAS, S.; MEINHARDT, W. Phase I Clinical and Pharmacokinetic Study of Kahalalide F in Patients with Advanced Androgen Refractory Prostate Cancer. Clinical Cancer Research, v.11, n.5, p.1854-1862, 2005.
RITU, B.; UMA, S.; NANDANA, S.; MONICA, J.; PAMITA, A.; MANPREET, K.; KUMAR, B. S.; GIRJESH, G. Structure of daunomycin complexed to d-TGATCA by two-dimensional nuclear magnetic resonance spectroscopy. European Journal of Medicinal Chemistry, v.41, n.1, p.27-39, 2006.
ROBERT, H.; FELING, G. O.; BUCHANAN, T. J.; MINCER, C. A.; KAUFFMAN, P. R.; FENICAL, W. Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angewandte Chemie, International Edition, v.42, n.3, p.355-357, 2003.
ROCHA, A. B.; LOPES, R. M.; SCHWARTSMANN, G. Natural products in anticancer therapy. Current Opinion in Pharmacology, v.1, n.4, p. 364-369, 2001.
SCHULTE, T. W.; NECKERS, L. M. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemotherapy and Pharmacology, v.42, n.4, p.273-279, 1998.
SENDEROWICZ, A. M. Flavopiridol: the First Cyclin-Dependent Kinase Inhibitor in Human Clinical Trials. Investigational New Drugs, v.17, n.3, p.313-320, 1999.
SOLAR, P.; HORVATH, V.; KLEBAN, J.; KOVAL, J.; SOLAROVA, Z.; KOZUBIK, A.; FEODOROCKO, P. Hsp90 inhibitor Geldanamycin increases the sensitivity of resistant ovarian adenocarcinoma cell line A2780cis to cisplatin. Neoplasma, v.54, n.2, p.127-130, 2007.
SPANO, A.; MONACO, G.; BARNI, S.; SCIOLA, L. Expression of cell kinetics and death during monocyte-macrophage differentiation: Effects of actinomycin D and vinblastine treatments. Histochemistry and Cell Biology, v.127, n.1, p.79-94, 2007.
SOUZA, M. V. N. New natural products able to act on the stabilization of microtubules, an important target against cancer. Química Nova, v.27, n.2, p.308-312, 2004.
STROSBERG, J. R.; KVOLS, L. K. A review of the current clinical trials for gastroenteropancreatic neuroendocrine tumours. Expert Opinion on Investigational Drugs, v.16, n.2, p.219-224, 2007.
SUN, M.; ALKON, D. L. Bryostatin-1: Pharmacology and Therapeutic Potential as a CNS Drug. CNS Drug Reviews, v.12, n.1, p.1-8, 2006.
TEDESCHI, A.; MONTILLO, M.; STROCCHI, E.; CAFRO, A. M.; TRESOLDI, E.; INTROPIDO, L.; NICHELATTI, M.; MARBELLO, L.; BARATE, C.; CAMAGGI, C. M.; MORRA, E. High-dose idarubicin in combination with Ara-C in patients with relapsed or refractory acute lymphoblastic leukemia: a pharmacokinetic and clinical study. Cancer Chemotherapy and Pharmacology, v.59, n.6, p.771-779, 2007.
THUN, M. J.; JEMAL, A.; SIEGEL, R.; WARD, E.; MURRAY, T.; XU, J.; SMIGAL, C. Cancer Statistics. CA-A Cancer Journal for Clinicians, v.56, n.2, p.106-130,2006.
TOKARSKA-SCHLATTNER, M.; ZAUGG, M.; ZUPPINGER, C.; WALLIMANN, T.; SCHLATTNER, U. New insights into doxorubicin-induced cardiotoxicity: The critical role of cellular energetics Journal of Molecular and Cellular Cardiology, v.41, n.3, p.389-405, 2006.
VARTERASIAN, M. L.; MOHAMMAD, R. M.; EILENDER, D. S.; HULBURD, K.; RODRIGUEZ, D. H.; PEMBERTON, P. A.; PLUDA, J. M.; DAN, M. D.; PETTIT, G. R.; CHEN, B. D.; AL-KATIB, A. M. Phase I study of bryostatin 1 in patients with relapsed non-Hodgkin’s lymphoma and chronic lymphocytic leukemia Journal of Clinical Oncology, v.16, n.1, p.56-62, 1998.
VELDHUIZEN, P. J. V.; FAULKNER, J. R.; LARAJR. P. N.; GUMERLOCK, P. H.; GOODWIN, J. W.; DAKHIL, S. R.; GROSS, H. M.; FLANIGAN R. C.;CRAWFORD E. D. Resistance to endocrine therapy in breast cancer. Cancer Chemotherapy and Pharmacology, v.56, p.39-46, 2005.
VENKAT, R.; MACHERLA, S. M.; RAMA RAO MANAM, R. R.; REED, K. A.; CHAO, T.; NICHOLSON, B.; DEYANAT-YAZDI, G. Structure-Activity Relationship Studies of Salinosporamide A (NPI-0052), a Novel Marine Derived Proteasome Inhibitor. Journal of Medicinal Chemistry, v.48, n.11, p.3684-3687, 2005.
XIE, X.; WATANABE, K.; WOJCICKI, W. A.; WANG, C. C. C.; TANG, Y. Biosynthesis of Lovastatin Analogs with a Broadly Specific Acyltransferase. Chemistry & Biology, v.13, n.11, p.1161-1169, 2006.
WHO: WORLD HEALTH ORGANIZATION. http://www.who.int/mediacentre/factsheets/fs297/en/index.html; Acessado em 15/04/2007.
ZANGARI, M.; CAVALLO, F.; TRICOT, G. Farnesyltransferase Inhibitors and Rapamycin in the Treatment of Multiple Myeloma. Current Pharmaceutical Biotechnology, v.7, n.6, p.449-453, 2006.